
Abstract
Rationale and Objective
Acute myocardial infarction (AMI) results in the recruitment of leukocytes to injured myocardium. Additionally, myocardium remote to the infarct zone also becomes inflamed and is associated with adverse left ventricular remodelling. Renal ischaemic syndromes have been associated with remote organ inflammation and impaired function. Here, we tested the hypothesis that AMI results in remote organ (renal) inflammation.
Methods
Mice were subjected to either AMI, sham procedure or no procedure and the inflammatory response in peripheral blood, injured and remote myocardium, and kidneys was studied at 24 h.
Results
AMI resulted in increased circulating neutrophils (P < 0.001) and monocytes (P < 0.001). mRNA for inflammatory mediators significantly increased in infarcted myocardium and in remote myocardium. VCAM-1 mRNA was increased in both infarcted and remote myocardium. VCAM-1 protein was also increased in the kidneys of AMI mice (P < 0.05) and immunofluorescence revealed localisation of VCAM-1 to glomeruli, associated with leukocyte infiltration and increased local inflammatory mRNA expression.
Conclusions
We conclude that in addition to local inflammation, AMI results in remote organ inflammation evidenced by (1) increased expression of mRNA for inflammatory cytokines, (2) marked upregulation of VCAM-1 in renal glomeruli, and (3) the recruitment and infiltration of leukocytes in the kidney.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myocardial infarction (AMI) results in the activation of the acute phase response [1, 2] and mobilisation and recruitment of leukocytes to the site of infarcted myocardium [3, 4]. Furthermore, myocardium that is remote from ischaemic zones has also been associated with the activation of pro-inflammatory pathways and infiltration of leukocytes [5], responses which are increasingly recognised as important in post-infarct left ventricular (LV) remodelling [2]. Renal ischemia syndromes have been associated with remote organ inflammation [6–9] including in the lungs [10] and brain [11, 12]. Substantial myocardial infarction is associated with multiple organ dysfunction, raising the possibility that activation of inflammatory pathways may extend beyond the heart to other critical organs.
Endothelial activation, marked by the expression of specific adhesion molecules, promotes leukocyte recruitment. For instance, upregulation of vascular cell adhesion molecule-1 (VCAM-1) is observed in the contralateral kidney in a mouse model of unilateral kidney ischaemia [13] and in remote myocardium of experimental models of AMI [5], whilst other adhesion molecules (e.g., intercellular adhesion molecule 1 (ICAM-1)) which are constitutively expressed remain unchanged [14]. Infiltrating leukocytes are a source of cytokines, such as IL-6, TNF-α and IL-1, which are involved in both the initial cellular inflammatory response and also subsequent tissue repair and fibrosis after injury [15]. IL1RN, a member of the IL-1 cytokine family, inhibits the activities of IL-1 and has been shown in clinical studies to be associated with higher plasma levels of C reactive protein (CRP) and IL-6 [16]. IL1R2 is a regulator of innate immunity and is the predominant IL-1 binding protein found in monocytes, neutrophils and B cells. It acts as a decoy receptor by sequestering IL-1 (which induces its expression) in the serum and tissues, thus preventing its downstream effects [17]. IL1R2 is significantly upregulated remotely in the lungs (where it is associated with inflammation and impaired function) in a murine renal ischaemia model [6]. IL-1 pathways are of particular interest since IL-1 antagonists (e.g. Anakinra, Canakinumab) are currently being studied in clinical trials [18, 19] in the context of acute myocardial infarction.
The inflammatory response resulting from AMI is systemic, with increased levels of soluble circulating inflammatory markers in humans and experimental models [20], and with the degree of cytokine release positively correlated with adverse outcomes [21]. TNF-α is strongly associated with the occurrence of reperfusion injury after recanalization [22] of the infarct-related artery and increased risk of mortality [23]. TNF-α, IL-6 and IL-1 are significantly elevated in patients with AMI complicated by cardiogenic shock when compared to patients with uncomplicated AMI [24]. IL-1 and IL-6 levels that are elevated in the first 2 days of hospitalization in unstable angina are associated with an increased risk of in-hospital coronary events [25]. Furthermore, cardio-renal syndromes type 1 and type 3, where acute dysfunction in the heart induces decreased kidney function and vice versa, are increasingly recognised but poorly understood [26, 27]. Acute kidney injury (AKI) is common in patients hospitalised with AMI and occurs in approximately one in five patients [28, 29]. Even the development of relatively mild renal impairment in AMI is associated with increased mortality [30–32] and morbidity including development of permanent renal impairment, progression to end-stage renal failure (ESRF) [30], increased length of hospital stay and cost [33].
Whilst the effects of acute kidney ischaemia have been shown to result in a pro-inflammatory cascade and release of cellular and soluble mediators with local [13] and systemic implications [6–12], there are no in vivo animal studies specifically addressing the effects of myocardial ischaemia on the kidneys acutely (i.e. in the first 24 h). Here, we tested the hypothesis that acute myocardial infarction results in remote organ (renal) inflammation, using a mouse model of myocardial infarction to study the inflammatory response in peripheral blood. To do so, we compared injured and remote myocardium and kidneys of mice that received ischaemic injury with those who received sham operation.
Materials and methods
Ethics statement
This study was undertaken with the approval of the University of Oxford Clinical Medicine Ethical Review Committee and procedures were conducted in accordance with the UK Home Office Animals (Science Procedures) Act 1986 (HMSO UK).
Animals
Female C57BL/6 J mice were purchased from Harlan (Blackthorn, United Kingdom) at 16 weeks of age and were housed in ventilated cages with a 12-h light/dark cycle and controlled temperature (20–22 °C), fed normal chow and given water ad libitum. All experiments were conducted on the mice at 24 weeks of age (n = 12–15/group). All surgery was performed under isoflurane anaesthesia, and all efforts were made to minimize suffering. Mice were sacrificed by exsanguination under terminal anaesthesia.
Mouse model of acute myocardial infarction
Following induction of anaesthesia with isoflurane, mice received 0.27 mg of buprenorphine and ventilated with a tidal volume 250 μL and respiratory rate 150/min (Hugo-Sachs MiniVent type 845, Harvard Apparatus, UK). Anaesthesia was maintained using 2 % isoflurane in 100 % oxygen. A left thoracotomy was performed in the fourth intercostal space; the left lung was deflated using a small piece of gauze soaked in saline. The pericardium was then removed and an intra-myocardial ligature placed 1–2 mm below the atrioventricular groove using a 6-0 polyethylene suture (Ethicon, UK). Lungs were then reinflated before the thorax was closed. Sham mice underwent the same protocol with the exception of the ligation of the coronary artery. Baseline (non-operated) mice underwent no operative procedure.
Murine transthoracic echocardiography
At 24 h following surgery, anaesthesia was induced and maintained with 2 % isoflurane and 98 % oxygen. Development of acute myocardial infarction following surgery was confirmed visually intra-operatively by the absence of blood flow to the anterior myocardial wall and at day 1 following surgery with the presence of a regional wall motion abnormality on transthoracic echocardiography. Transthoracic echocardiography was carried out with a Visualsonics Vevo 2100 ultrasound system (Amsterdam, The Netherlands). Fractional shortening percentage was calculated in both parasternal and short axis views to assess myocardial function/size of infarct using the following formula: end-diastolic diameter − end-systolic diameter/end-diastolic diameter × 100.
Flow cytometry
Whole blood was obtained following terminal anaesthesia via left ventricular puncture (n = 5/group). Red blood cells were lysed with lysis buffer (eBioscience, San Diego, USA). Following washing in fluorescent activated cell sorting (FACS) buffer [phosphate buffered saline (PBS), 1 mM ethylenediaminetetraacetic acid (EDTA) and 1 % bovine serum albumin (BSA)], cells were incubated with a Live/Dead cell viability stain (Invitrogen) for 30 min at room temperature. Cells were then incubated with anti-FcgRII/RIII blocking antibody (BD Pharmingen, Oxford, UK) for 15 min and then incubated with the following antibodies: CD11b-APC, Ly6G-PE (both BD Pharmingen) and CD115-PerCP (eBioscience). Cells were fixed in 1 % PFA and analysed using the flow cytometer (CyAN ADP Flow cytometer, Dako, Ely, UK). Data were then analysed using FlowJo software, version 7.6.3 (Tree Star Inc, Oregon, USA), and compensation and gates were set using single-stain controls, IgG controls and fluorescence minus one.
RNA extraction and quantitative RT-PCR
Myocardium and kidney tissue were both mechanically homogenised in TRIzol and RNA was extracted with chloroform and ethanol (n = 6–8/group) and purified using an RNeasy mini kit (Qiagen, Crawley, UK) as per the manufacturer’s instructions. 1 μg of total RNA was reversed transcribed using a QuantiTect Reverse Transcription kit with oligo dTs and random hexamers as primers. Real-time PCR was carried out with 1 μl of cDNA in a 10 μl reaction mix using Sybr Green Gene Expression assays (Applied Biosystems, Warrington UK) and the primers as outlined in Table 1. Quantification was performed by the 2−ΔΔCT method [34], normalised to the housekeeping gene cyclophilin.
Western blot
Protein lysates were obtained following mechanical homogenization of fresh kidney tissue in cell lysis reagent for mammalian tissues (Sigma, UK) (n = 6/group). Samples were separated by SDS-PAGE (4–12 % Bis/Tris resolving gel, Invitrogen, Paisley, UK) and transferred to polyvinylidene difluoride (PVDF) membranes using the iBlot® dry blotting system (Invitrogen, Paisley, UK). Immunoblotting was performed using VCAM-1 antibody (R&D Systems, Abingdon, UK). Immunoreactive bands were visualised by chemiluminescence (GE Healthcare Life Sciences, Amersham, UK). Signal intensity was analysed using ImagePro Plus version 6.1 (MediaCybernetics, Silver Spring, MD) and normalised to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Histology
For assessment of myocardial infiltrating leukocytes, haematoxylin and eosin (H&E) staining was performed. At 24 h after myocardial infarction, mice were exsanguinated and immediately perfused with phosphate buffered saline (PBS), and their hearts were harvested and fixed in 4 % paraformaldehyde and embedded in paraffin. Sections (5 μm) were stained with H&E reagents for histological examination. Ten random fields of view (FOV) were obtained at 40 × magnification on light microscopy at mid-cavity level of the left ventricle. The infarct zone was selected anatomically as the anterior wall (as confirmed on transthoracic echocardiography by the presence of a regional wall abnormality) with the remote myocardium selected as the contralateral wall. Infiltrating leukocytes were identified based on their morphological appearance and counted (n = 4/group).
Immunohistochemistry
After the mice were sacrificed as described above, their kidneys were embedded in optimal cutting temperature (OCT) compound. Sections (8 μm) were incubated with a rat anti-mouse CD11b antibody (BD biosciences, UK) and then an anti-rat secondary antibody conjugated to Alexa-fluor 750. Images were obtained using a LSM510 Meta confocal system (Zeiss, UK). Ten random FOV were obtained for each kidney (n = 6/group) and positive cells were analysed using ImagePro Plus version 6.1 (MediaCybernetics, Silver Spring, MD). Other sections were incubated with a rat anti-mouse VCAM-1 primary antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA) alone or in combination with a goat anti-mouse PECAM-1 primary antibody (R&D Systems, Minneapolis, MN, USA). An anti-rat secondary antibody conjugated to Alexa-fluor 750 was used to detect VCAM-1 and an anti-goat secondary antibody conjugated to Alexa-fluor 488 to detect PECAM-1. Images were obtained and analysed as described above. Unstained and secondary antibody-only stained sections served as negative controls and IgG controls were conducted to exclude non-specific binding.
Evaluating renal function
Plasma was obtained by centrifugation of whole blood. Serum creatinine levels were measured using a mouse creatinine assay kit (n = 4/group) (Chrystal Chem, Inc., IL, USA).
Statistics
Data are expressed as mean and SEM and tested for significance using Student’s t test or one-way ANOVA. All statistics were carried out using GraphPad Prism software (La Jolla, CA, USA). A two-tailed P value of < 0.05 was considered statistically significant.
Results
Acute myocardial infarction
As expected, fractional shortening (FS) was significantly reduced in mice subjected to myocardial infarction, compared to both sham-operated and non-operated animals at day 1 following surgery (P < 0.001; Fig. 1a–c). At day 1, MI results in a significant leukocytosis with increases in circulating CD11b + Ly6G + neutrophils and CD11b + CD115 + Ly6G- monocytes (P < 0.001) in comparison to non-operated and sham-operated animals (Fig. 1d, e).

AMI results in impaired LV function, peripheral leukocytosis and infiltration of leukocytes to myocardium. M-mode echocardiographic measurements of left ventricular function through the mid-cavity of a sham-operated mouse (a) and an infarcted mouse (b) resulting in a significant reduction in left ventricular fractional shortening (c) (P < 0.001). AMI results in a significant systemic inflammatory response with a systemic leukocytosis (neutrophilia (red gate, P < 0.001) and monocytosis (green gate, P < 0.001)) (d, e). H&E staining of myocardium (× 40 magnification, scale bar 50 μm; inset × 100 magnification, scale bar 10 μm) reveals significant infiltration of leukocytes to infarcted myocardium in comparison with sham-operated hearts (black arrows, f) P < 0.001
Myocardial infarction results in local inflammation of the ischaemic zone
H&E staining revealed greatly increased numbers of leukocytes in the myocardial infarct zone, by 10.8 ± 3.24-fold (n = 4/group, P < 0.001, Fig. 1f). PCR of infarcted myocardium revealed upregulation of inflammatory mRNA expression of IL6 by 11 ± 0.33-fold (P < 0.01), TNF-α by 32.94 ± 2.34-fold (P < 0.01), IL1RN by 5.41 ± 0.24-fold (P < 0.05), and IL1R2 by 4.31 ± 0.98-fold (n = 8/group, P < 0.05). VCAM-1 mRNA expression in myocardium was also increased by 19.62 ± 1.11-fold (P < 0.01) (Fig. 2).

Myocardial mRNA expression of normal myocardium, infarcted myocardium and remote myocardium. In comparison to normal (baseline) mice there were significant increases in IL-6 (P < 0.01, P < 0.05), VCAM-1 (P < 0.01, P < 0.05) and TNF-α (P < 0.01, P < 0.05) in both the remote zones and infarct zones respectively at 24 h. Significant increases in IL1RN (P < 0.05) and IL1R2 (P < 0.05) in infarct zones in comparison to normal myocardium were also observed. Whilst there was a trend of increase in IL1RN and IL1R2 in remote myocardium, these did not reach statistical significance
Myocardial infarction results in inflammation of remote myocardium
In addition to investigating inflammation in the infarct zone, we investigated whether there was inflammation in myocardium that was remote from the ischaemically injured areas. H&E staining revealed significantly increased numbers of leukocytes in the remote myocardium by 3.9 ± 1.1-fold (n = 4/group, P < 0.01) (Fig. 1f) in comparison to baseline myocardium which was associated with upregulation of mRNA for inflammatory mediators. PCR revealed significant increases in mRNA expression of IL-6 by 5.01 ± 0.73-fold (P < 0.05), TNF-α by 15.18 ± 4.23-fold (P < 0.05) and VCAM-1 by 13.12 ± 2.15-fold (P < 0.05) in comparison to baseline (Fig. 2).
Remote inflammation in the kidney
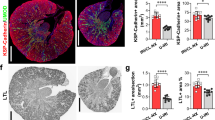
Having observed the upregulation of VCAM-1 in both the infarct zone and remote myocardium we proceeded to quantify VCAM-1 mRNA and protein in the kidney. Whilst VCAM-1 mRNA expression was not significantly elevated in the kidney at the time point examined (1.39 ± 0.51-fold; P = n.s.) (Fig. 5), VCAM-1 protein was significantly increased in kidneys of infarcted mice (Fig. 3b) in comparison to baseline and sham-operated mice by 1.81 ± 0.14-fold and 2.6 ± 0.27-fold, respectively. Immunofluorescence showed that VCAM-1 was not expressed under basal conditions but was upregulated and clearly localised to glomeruli in mice with myocardial infarction (Fig. 3a) and to the same cells expressing PECAM-1, indicating upregulation by glomerular endothelial cells (Fig. 3c).

Renal VCAM-1 is upregulated as a result of AMI. Renal VCAM-1 expression (green, DAPI blue) is significantly upregulated in kidneys of infarcted mice in comparison to baseline and sham-operated animals. The VCAM-1 was found to be exclusively localised to the glomeruli, a with the same cells also expressing the endothelial cell marker PECAM-1 (c). Renal VCAM-1 protein was significantly increased in comparison to uninfarcted and sham-operated animals (b) (P < 0.05). × 63 magnification, scale bar 50 μm, zoomed inset: scale bar 25 μm
Myocardial Infarction results in increased leukocyte infiltration to remote kidneys
In kidneys of mice with acute myocardial infarction we found that there was an increase in numbers of CD11b + leukocytes throughout the kidney by immunohistochemistry (Fig. 4). In contrast, in mice that did not undergo surgery and in sham-operated mice, only occasional CD11b + leukocytes were visualised.

AMI results in significant leukocyte recruitment to remote kidneys. AMI results in a significant increase in infiltrating CD11b + leukocytes (green, DAPI blue) to remote kidneys in comparison with baseline or sham-operated animals (white arrows (inset), P < 0.001). × 63 magnification, scale bar 50 μm, zoomed inset: scale bar 25 μm
Myocardial Infarction results in remote kidney inflammation
Having observed increases in both VCAM-1 and leukocyte infiltration in the kidneys, we next sought to investigate the effects of myocardial infarction on mRNA for other inflammatory mediators. Several inflammatory genes were also significantly upregulated in the kidneys of infarcted mice in comparison to baseline and sham-operated animals: IL6 by 2.87 ± 0.13-fold and 5.28 ± 0.07-fold (P < 0.001), TNF-α by 7.41 ± 1.8-fold and 3.67 ± 0.15-fold (P < 0.01), IL1RN by 3.86 ± 0.19-fold and 4.69 ± 0.28-fold (P < 0.01) and IL1R2 by 10.3 ± 0.33-fold and 14.57 ± 4.9-fold (P < 0.01) (n = 8/group) (Fig. 5).

Renal mRNA expression of baseline, sham-operated and infarcted animals. In comparison to baseline and sham-operated mice, IL-6 (P < 0.001), TNF-α (P < 0.05), IL1RN (P < 0.01) and IL1R2 (P < 0.01) were significantly increased. At 24 h, VCAM-1 mRNA expression was not found to be significantly different from baseline or sham-operated mice
Local inflammation in the acute setting has no effect on plasma creatinine
Acute myocardial infarction, did not affect plasma creatinine (baseline: 8.25 ± 0.3 μmol/l vs. MI: 10.33 ± 2.0 μmol/l, P = n.s.) despite evidence of renal inflammation.
Discussion
There is an increasing appreciation that systemic inflammatory pathways are activated in a variety of medical conditions (e.g. AMI, severe sepsis, trauma, renal ischaemia) and that these pathways play an important role in the development of remote organ inflammation leading to adverse outcomes. In experimental models of acute kidney injury (AKI), remote organ (heart [9, 35], lung [36], liver [8] and brain [12]) inflammation has been described, resulting in dysfunction. The acute effects of AMI (first 24 h) on remote organ inflammation have not previously been investigated. Here, using a mouse model we investigate the acute effects of AMI on inflammation remotely in the kidney. The present study demonstrates that: (1) AMI induces a local inflammatory response, characterised by infiltration of leukocytes and increased expression of mRNA for inflammatory cytokines; (2) cytokines are upregulated in myocardium that is remote from the site of ischaemic injury; (3) AMI is associated with systemic inflammation, reflected in increased peripheral blood neutrophils and monocytes; (4) AMI is also associated with remote organ inflammation evidenced by (a) increased expression of mRNA for inflammatory cytokines, (b) marked upregulation of VCAM-1 in renal glomeruli and (c) by the recruitment and infiltration of leukocytes throughout the kidney.
Infiltrating leukocytes to areas of acute myocardial inflammation and post-MI myocardium are a source of inflammatory cytokines (e.g. IL-6, IL-1 and TNF-α) and contribute to local inflammation, remodelling and fibrosis of the myocardium [37, 38]. It has recently been observed that leukocytes also infiltrate remote myocardium post-MI, as a consequence of increased expression of adhesion molecules and inflammatory cytokines, and may contribute to adverse left ventricular dilatation, fibrosis and impaired function [5].
De novo renal impairment or AKI in the setting of AMI affects 20 % of hospitalised patients [28, 29, 32]. The degree of impaired renal function is associated with increased short- and long-term mortality [30, 31, 39], and in this setting even mild transient renal dysfunction is an independent risk factor for long-term survival [28].
Upregulation of IL-6, IL-1 and TNF-α in the kidney may contribute to acute renal dysfunction and also subsequent fibrosis and chronic impairment [40, 41]. The expression of VCAM-1 exclusively in the glomeruli of the kidneys supports recent observations of glomerular VCAM-1 expression as a result of TNF-α stimulation in keeping with acute inflammation [42].
We found that this was associated with a significant increase in leukocyte infiltration which suggests that endothelial activation at remote sites may play a role in initiating or exacerbating renal inflammation and its sequelae with the same glomerular cells expressing PECAM-1 (Fig. 3c). However, glomerular mesangial cells may also play a role in VCAM-1 expression and monocyte infiltration [43].
There are a number of possible mechanisms that could explain these findings. Soluble mediators such as inflammatory cytokines (IL-6, TNF-α) released from ischaemic myocardium may act directly at remote areas. Studies have shown upregulation of mRNA expression of cytokines and resultant phenotypic evidence of injury associated with dysfunction [44] with blockade of these cytokines attenuating remote inflammation and injury in various ischaemic models [9, 45, 46]. Increased inflammatory cytokines result in increased apoptosis in cardiomyocytes [9] and, additionally, an increase in oxidative stress in hepatocytes as a consequence of AKI [7]. Remote organ inflammation may also result from cellular mediators both as part of the innate and adaptive immune response. The innate immune response may be activated by the activation of toll-like receptors (TLRs) and release of reactive oxygen species or by the activation of the complement pathway as a result of ischaemic tissue which in turn would lead to the release of cytokines and endothelial activation with trafficking of leukocytes [47]. Adaptive immunity may also be activated with T cells having been shown to play a central role in renal ischaemic models [48] in both the acute inflammatory response [49] and also long-term renal dysfunction [50]. In addition to cellular mediators, there also may be other key modulators, e.g., lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), which can serve as a pro-inflammatory adhesion molecule [51] and whose expression and activation can lead to the generation of reactive oxygen species [52]. Abrogation of LOX-1 in a mouse model has been shown to reduce inflammation, recruitment of leukocytes and the development of fibrosis in kidneys following AMI [41].
It is clear that the mechanisms governing remote organ inflammation are complex and may all contribute to different degrees in addition to contributions from other factors such as microparticles [53] and neurogenic pathways. Most of our current understanding results from renal ischaemic models with the effects of AMI on remote organs and the underlying mechanisms for this remaining poorly characterised.
A major current limitation in clinical practice is the absence of a biomarker that is sensitive enough to detect small changes in renal function. Routinely used measurements such as serum creatinine or blood urea nitrogen start to rise only after a significant reduction in renal function, though novel biomarkers such as kidney injury molecule-1 (Kim-1) and lipocalin-2 (Lcn2) may hold some promise for the future [54]. Renal expression of VCAM-1 could potentially be used as a biomarker for renal inflammation post-MI to identify patients at risk of AKI in this scenario [13].
In view of these findings, there may be a role of emerging drugs in this patient group with drugs such as IL-1 antagonists (e.g. Anakinra, Canakinumab). Indeed, interleukin-1 blockade in a mouse model of AMI [55] and in a small clinical pilot study has been found to be safe in AMI and favourably affected left ventricular remodelling [18]. Thus these drugs may have a role in the management of AKI in AMI, resulting in improvements in short-term AKI and also in the development of long-term renal fibrosis, ultimately improving long-term mortality and morbidity.
There are limitations to this study. A correlation between the magnitude of myocardial inflammation and the extent of remote organ inflammation could be beneficial in identifying patients at higher risk of developing remote organ injury at the bedside. However, due to small numbers and the marked variability of the magnitude of inflammation, we had insufficient numbers for correlation analysis. Additionally, we were unable to demonstrate functional significance of remote kidney inflammation: creatinine (as used here) is an insensitive marker for glomerular filtration rate (GFR), however there are currently no better measures available. Finally, poor cardiac function and therefore renal hypoperfusion may account for some of our findings and the sham surgery group does not adequately control for this variable. However, in similar experimental models, the blood pressure reduction following AMI has only been found to be modest [41], and in other ischaemic models (e.g. renal ischaemia) remote organ inflammation has been found in the absence of any change in cardiac function [7–10], suggesting that blood pressure would not be the only mechanism to explain our findings.
In summary, this study demonstrates remote kidney inflammation at 24 h following AMI, characterised by endothelial activation, recruitment of infiltrating leukocytes and the release of local inflammatory mediators. On the basis of these findings, further work is merited to establish the time course of renal injury following AMI and the effect of remote kidney inflammation on function.
References
Kushner I, Broder ML, Karp D. Control of the acute phase response. Serum C-reactive protein kinetics after acute myocardial infarction. J Clin Invest. 1978;61(2):235–42.
Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53(1):31–47.
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–47.
Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–6.
Lee WW, Marinelli B, van der Laan AM, Sena BF, Gorbatov R, Leuschner F, et al. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol. 2012;59(2):153–63.
Grigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, Rabb H. The local and systemic inflammatory transcriptome after acute kidney injury. J Am Soc Nephrol. 2008;19(3):547–58.
Golab F, Kadkhodaee M, Zahmatkesh M, Hedayati M, Arab H, Schuster R, et al. Ischemic and non-ischemic acute kidney injury cause hepatic damage. Kidney Int. 2009;75(8):783–92.
Park SW, Chen SW, Kim M, Brown KM, Kolls JK, D’Agati VD, et al. Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab Invest. 2011;91(1):63–84.
Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003;14(6):1549–58.
Kramer AA, Postler G, Salhab KF, Mendez C, Carey LC, Rabb H. Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int. 1999;55(6):2362–7.
Adachi N, Lei B, Deshpande G, Seyfried FJ, Shimizu I, Nagaro T, et al. Uraemia suppresses central dopaminergic metabolism and impairs motor activity in rats. Intensive Care Med. 2001;27(10):1655–60.
Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, et al. Acute kidney injury leads to inflammation and functional changes in the brain. J Am Soc Nephrol. 2008;19(7):1360–70.
Akhtar AM, Schneider JE, Chapman SJ, Jefferson A, Digby JE, Mankia K, et al. In vivo quantification of VCAM-1 expression in renal ischemia reperfusion injury using non-invasive magnetic resonance molecular imaging. PLoS ONE. 2010;5(9):e12800.
Nakashima Y, Raines EW, Plump AS, Breslow JL, Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol. 1998;18(5):842–51.
Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94(12):1543–53.
Reiner AP, Wurfel MM, Lange LA, Carlson CS, Nord AS, Carty CL, et al. Polymorphisms of the IL1-receptor antagonist gene (IL1RN) are associated with multiple markers of systemic inflammation. Arterioscler Thromb Vasc Biol. 2008;28(7):1407–12.
Mantovani A, Bonecchi R, Martinez FO, Galliera E, Perrier P, Allavena P, et al. Tuning of innate immunity and polarized responses by decoy receptors. Int Arch Allergy Immunol. 2003;132(2):109–15.
Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am J Cardiol. 2010;105(10):1371–1377e1.
Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Am Heart J. 2011;162(4):597–605.
Balbay Y, Tikiz H, Baptiste RJ, Ayaz S, Sasmaz H, Korkmaz S. Circulating interleukin-1 beta, interleukin-6, tumor necrosis factor-alpha, and soluble ICAM-1 in patients with chronic stable angina and myocardial infarction. Angiology. 2001;52(2):109–14.
Valgimigli M, Ceconi C, Malagutti P, Merli E, Soukhomovskaia O, Francolini G, et al. Tumor necrosis factor-alpha receptor 1 is a major predictor of mortality and new-onset heart failure in patients with acute myocardial infarction: the Cytokine-Activation and Long-Term Prognosis in Myocardial Infarction (C-ALPHA) study. Circulation. 2005;111(7):863–70.
Blancke F, Claeys MJ, Jorens P, Vermeiren G, Bosmans J, Wuyts FL, et al. Systemic inflammation and reperfusion injury in patients with acute myocardial infarction. Mediators Inflamm. 2005;2005(6):385–9.
Kohsaka S, Menon V, Lowe AM, Lange M, Dzavik V, Sleeper LA, et al. Systemic inflammatory response syndrome after acute myocardial infarction complicated by cardiogenic shock. Arch Intern Med. 2005;165(14):1643–50.
Debrunner M, Schuiki E, Minder E, Straumann E, Naegeli B, Mury R, et al. Proinflammatory cytokines in acute myocardial infarction with and without cardiogenic shock. Clin Res Cardiol. 2008;97(5):298–305.
Biasucci LM, Liuzzo G, Fantuzzi G, Caligiuri G, Rebuzzi AG, Ginnetti F, et al. Increasing levels of interleukin (IL)-1Ra and IL-6 during the first 2 days of hospitalization in unstable angina are associated with increased risk of in-hospital coronary events. Circulation. 1999;99(16):2079–84.
Hillege HL, van Gilst WH, van Veldhuisen DJ, Navis G, Grobbee DE, de Graeff PA, et al. Accelerated decline and prognostic impact of renal function after myocardial infarction and the benefits of ACE inhibition: the CATS randomized trial. Eur Heart J. 2003;24(5):412–20.
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52(19):1527–39.
Parikh CR, Coca SG, Wang Y, Masoudi FA, Krumholz HM. Long-term prognosis of acute kidney injury after acute myocardial infarction. Arch Intern Med. 2008;168(9):987–95.
Amin AP, Salisbury AC, McCullough PA, Gosch K, Spertus JA, Venkitachalam L, et al. Trends in the incidence of acute kidney injury in patients hospitalized with acute myocardial infarction. Arch Intern Med. 2012;172(3):246–53.
Newsome BB, Warnock DG, McClellan WM, Herzog CA, Kiefe CI, Eggers PW, et al. Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Intern Med. 2008;168(6):609–16.
Aengus Murphy C, Robb SD, Weir RA, McDonagh TA, Dargie HJ. Declining renal function after myocardial infarction predicts poorer long-term outcome. Eur J Cardiovasc Prev Rehabil. 2010;17(2):181–6.
Bruetto RG, Rodrigues FB, Torres US, Otaviano AP, Zanetta DM, Burdmann EA. Renal function at hospital admission and mortality due to acute kidney injury after myocardial infarction. PLoS ONE. 2012;7(4):e35496.
Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16(11):3365–70. doi:10.1681/ASN.2004090740. pii:ASN.2004090740.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45.
Nath KA, Grande JP, Croatt AJ, Frank E, Caplice NM, Hebbel RP, et al. Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol. 2005;166(4):963–72.
Hoke TS, Douglas IS, Klein CL, He Z, Fang W, Thurman JM, et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol. 2007;18(1):155–64.
Turner NA, Das A, Warburton P, O’Regan DJ, Ball SG, Porter KE. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am J Physiol Heart Circ Physiol. 2009;297(3):H1117–27.
Hori M, Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc Res. 2009;81(3):457–64.
Gibson CM, Pinto DS, Murphy SA, Morrow DA, Hobbach HP, Wiviott SD, et al. Association of creatinine and creatinine clearance on presentation in acute myocardial infarction with subsequent mortality. J Am Coll Cardiol. 2003;42(9):1535–43.
Lekawanvijit S, Kompa AR, Zhang Y, Wang BH, Kelly DJ, Krum H. Myocardial Infarction Impairs Renal Function, Induces Renal Interstitial Fibrosis and Increases renal KIM-1 Expression: Implications for Cardiorenal Syndrome. Am J Physiol Heart Circ Physiol. 2012;302(9):H1884–93.
Lu J, Wang X, Wang W, Muniyappa H, Deshmukh A, Hu C, et al. Abrogation of lectin-like oxidized LDL receptor-1 attenuates acute myocardial ischemia-induced renal dysfunction by modulating systemic and local inflammation. Kidney Int. 2012;82(4):436–44.
Asgeirsdottir SA, van Solingen C, Kurniati NF, Zwiers PJ, Heeringa P, van Meurs M, et al. MicroRNA-126 contributes to renal microvascular heterogeneity of VCAM-1 protein expression in acute inflammation. Am J Physiol Renal Physiol. 2012;302(12):F1630–9.
Wuthrich RP. Vascular cell adhesion molecule-1 (VCAM-1) expression in murine lupus nephritis. Kidney Int. 1992;42(4):903–14.
Hassoun HT, Grigoryev DN, Lie ML, Liu M, Cheadle C, Tuder RM, et al. Ischemic acute kidney injury induces a distant organ functional and genomic response distinguishable from bilateral nephrectomy. Am J Physiol Renal Physiol. 2007;293(1):F30–40.
Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int. 2008;74(7):901–9.
Tracz MJ, Juncos JP, Croatt AJ, Ackerman AW, Grande JP, Knutson KL, et al. Deficiency of heme oxygenase-1 impairs renal hemodynamics and exaggerates systemic inflammatory responses to renal ischemia. Kidney Int. 2007;72(9):1073–80.
Chakraborti T, Mandal A, Mandal M, Das S, Chakraborti S. Complement activation in heart diseases. Role of oxidants. Cell Signal. 2000;12(9–10):607–17.
Burne-Taney MJ, Kofler J, Yokota N, Weisfeldt M, Traystman RJ, Rabb H. Acute renal failure after whole body ischemia is characterized by inflammation and T cell-mediated injury. Am J Physiol Renal Physiol. 2003;285(1):F87–94.
Satpute SR, Park JM, Jang HR, Agreda P, Liu M, Gandolfo MT, et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J Immunol. 2009;183(2):984–92.
Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75(5):526–35.
Honjo M, Nakamura K, Yamashiro K, Kiryu J, Tanihara H, McEvoy LM, et al. Lectin-like oxidized LDL receptor-1 is a cell-adhesion molecule involved in endotoxin-induced inflammation. Proc Natl Acad Sci USA. 2003;100(3):1274–9.
Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, et al. Critical role of the NAD(P)H oxidase subunit p47phox for left ventricular remodeling/dysfunction and survival after myocardial infarction. Circ Res. 2007;100(6):894–903.
Porto I, Biasucci LM, De Maria GL, Leone AM, Niccoli G, Burzotta F, et al. Intracoronary microparticles and microvascular obstruction in patients with ST elevation myocardial infarction undergoing primary percutaneous intervention. Eur Heart J. 2012;33(23):2928–38.
Hoffmann D, Adler M, Vaidya VS, Rached E, Mulrane L, Gallagher WM, et al. Performance of novel kidney biomarkers in preclinical toxicity studies. Toxicol Sci. 2010;116(1):8–22.
Abbate A, Van Tassell BW, Seropian IM, Toldo S, Robati R, Varma A, et al. Interleukin-1beta modulation using a genetically engineered antibody prevents adverse cardiac remodelling following acute myocardial infarction in the mouse. Eur J Heart Fail. 2010;12(4):319–22.
Acknowledgments
Phil Townsend and James Brown are gratefully acknowledged for general laboratory management. The authors thank BMS staff for their expert care of mice used in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Graham Wallace
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ruparelia, N., Digby, J.E., Jefferson, A. et al. Myocardial infarction causes inflammation and leukocyte recruitment at remote sites in the myocardium and in the renal glomerulus. Inflamm. Res. 62, 515–525 (2013). https://doi.org/10.1007/s00011-013-0605-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-013-0605-4