

Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination
- PMID: 9847317
- PMCID: PMC103818
- DOI: 10.1128/JVI.73.1.152-160.1999
Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination
Abstract
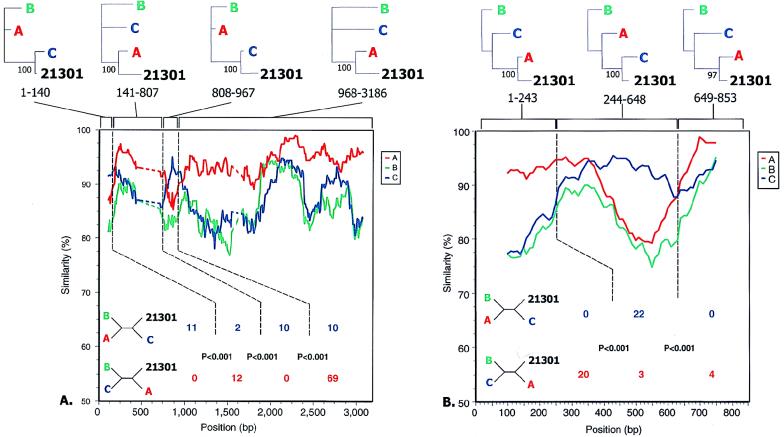
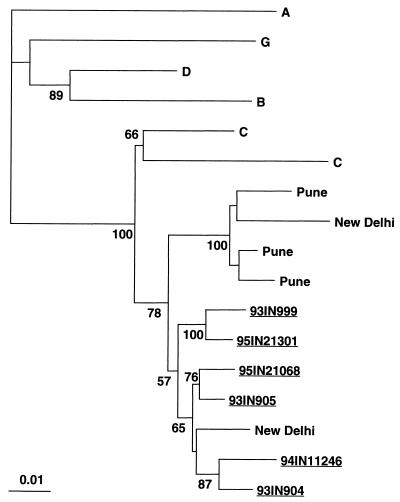
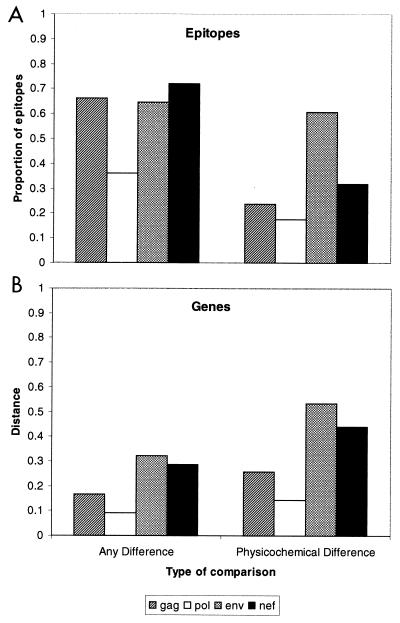
The development of an effective human immunodeficiency virus type 1 (HIV-1) vaccine is likely to depend on knowledge of circulating variants of genes other than the commonly sequenced gag and env genes. In addition, full-genome data are particularly limited for HIV-1 subtype C, currently the most commonly transmitted subtype in India and worldwide. Likewise, little is known about sequence variation of HIV-1 in India, the country facing the largest burden of HIV worldwide. Therefore, the objective of this study was to clone and characterize the complete genome of HIV-1 from seroconverters infected with subtype C variants in India. Cocultured HIV-1 isolates were obtained from six seroincident individuals from Pune, India, and virtually full-length HIV-1 genomes were amplified, cloned, and sequenced from each. Sequence analysis revealed that five of the six genomes were of subtype C, while one was a mosaic of subtypes A and C, with multiple breakpoints in env, nef, and the 3' long terminal repeat as determined by both maximal chi2 analysis and phylogenetic bootstrapping. Sequences were compared for preservation of known cytotoxic T lymphocyte (CTL) epitopes. Compared with those of the HIV-1LAI sequence, 38% of well-defined CTL epitopes were identical. The proportion of nonconservative substitutions for Env, at 61%, was higher (P < 0.001) than those for Gag (24%), Pol (18%), and Nef (32%). Therefore, characterized CTL epitopes demonstrated substantial differences from subtype B laboratory strains, which were most pronounced in Env. Because these clones were obtained from Indian seroconverters, they are likely to facilitate vaccine-related efforts in India by providing potential antigens for vaccine candidates as well as for assays of vaccine responsiveness.
Figures



Similar articles
-
Characterization of a virtually full-length human immunodeficiency virus type 1 genome of a prevalent intersubtype (C/B') recombinant strain in China.J Virol. 2000 Dec;74(23):11367-76. doi: 10.1128/jvi.74.23.11367-11376.2000. J Virol. 2000. PMID: 11070037 Free PMC article.
-
The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin.J Virol. 1996 Oct;70(10):7013-29. doi: 10.1128/JVI.70.10.7013-7029.1996. J Virol. 1996. PMID: 8794346 Free PMC article.
-
Sequence and drug susceptibility of subtype C reverse transcriptase from human immunodeficiency virus type 1 seroconverters in Zimbabwe.J Virol. 1997 Jul;71(7):5441-8. doi: 10.1128/JVI.71.7.5441-5448.1997. J Virol. 1997. PMID: 9188616 Free PMC article.
-
Near full-length clones and reference sequences for subtype C isolates of HIV type 1 from three different continents.AIDS Res Hum Retroviruses. 2001 Jan 20;17(2):161-8. doi: 10.1089/08892220150217247. AIDS Res Hum Retroviruses. 2001. PMID: 11177395
-
Rapid, complex adaptation of transmitted HIV-1 full-length genomes in subtype C-infected individuals with differing disease progression.AIDS. 2013 Feb 20;27(4):507-18. doi: 10.1097/QAD.0b013e32835cab64. AIDS. 2013. PMID: 23370465 Free PMC article.
Cited by
-
Comparative genomics reveals insights into genetic variability and molecular evolution among sugarcane yellow leaf virus populations.Sci Rep. 2021 Mar 30;11(1):7149. doi: 10.1038/s41598-021-86472-z. Sci Rep. 2021. PMID: 33785787 Free PMC article.
-
Epidemiological Surveillance of HIV-1 Transmitted Drug Resistance in Spain in 2004-2012: Relevance of Transmission Clusters in the Propagation of Resistance Mutations.PLoS One. 2015 May 26;10(5):e0125699. doi: 10.1371/journal.pone.0125699. eCollection 2015. PLoS One. 2015. PMID: 26010948 Free PMC article.
-
Local Virus Extinctions following a Host Population Bottleneck.J Virol. 2015 Aug;89(16):8152-61. doi: 10.1128/JVI.00671-15. Epub 2015 May 27. J Virol. 2015. PMID: 26018153 Free PMC article.
-
Comparative genomics of Korean infectious bronchitis viruses (IBVs) and an animal model to evaluate pathogenicity of IBVs to the reproductive organs.Viruses. 2012 Oct 30;4(11):2670-83. doi: 10.3390/v4112670. Viruses. 2012. PMID: 23202499 Free PMC article.
-
Recombination of Globally Circulating Varicella-Zoster Virus.J Virol. 2015 Jul;89(14):7133-46. doi: 10.1128/JVI.00437-15. J Virol. 2015. PMID: 25926648 Free PMC article.
References
-
- Anonymous. Global AIDS surveillance. WHO Weekly Epidemiological Record. 1993;68(27):195–196. - PubMed
-
- Anonymous. World Health Report. Geneva, Switzerland: World Health Organization; 1997.
-
- Bachmann M H, Delwart E L, Shpaer E G, Lingenfelter P, Singal R, Mullins J I. Rapid genetic characterization of HIV type 1 strains from four World Health Organization-sponsored vaccine evaluation sites using a heteroduplex mobility assay. WHO Network for HIV isolation and characterization. AIDS Res Hum Retroviruses. 1994;10:1345–1353. - PubMed
-
- Baskar P V, Ray S C, Rao R, Quinn T C, Hildreth J E, Bollinger R C. Presence in India of HIV type 1 similar to North American strains. AIDS Res Hum Retroviruses. 1994;10:1039–1041. - PubMed
-
- Bollinger R C, Brookmeyer R S, Mehendale S M, Paranjape R S, Shepherd M E, Gadkari D A, Quinn T C. Risk factors and clinical presentation of acute primary HIV infection in India. JAMA. 1997;278:2085–2089. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
