

doi: 10.1073/pnas.95.2.681.
Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis
Affiliations
- PMID: 9435252
- PMCID: PMC18480
- DOI: 10.1073/pnas.95.2.681
Item in Clipboard
Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis
Proc Natl Acad Sci U S A.
.
Abstract
Nonsteroidal antiinflammatory drugs (NSAIDs) can inhibit colorectal tumorigenesis and are among the few agents known to be useful for the chemoprevention of neoplasia. Here, we show that the tumor suppressive effects of NSAIDs are not likely to be related to a reduction in prostaglandins but rather are due to the elevation of the prostaglandin precursor arachidonic acid (AA). NSAID treatment of colon tumor cells results in a dramatic increase in AA that in turn stimulates the conversion of sphingomyelin to ceramide, a known mediator of apoptosis. These results have significant implications for understanding and improving colon cancer chemoprevention.
Figures
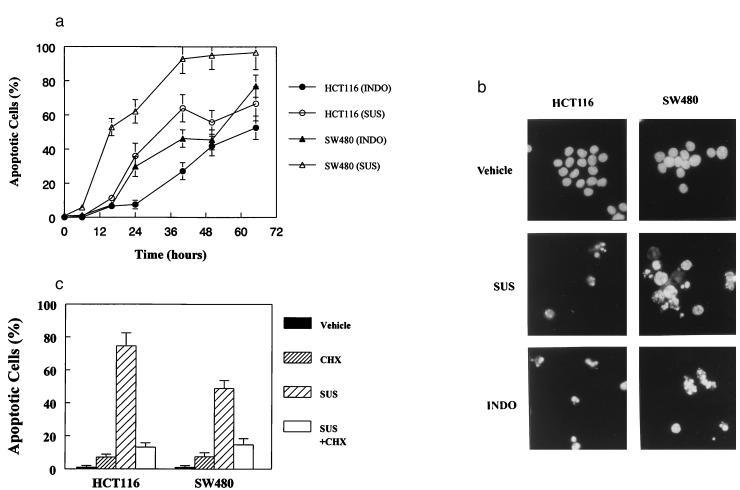
NSAIDs induce apoptosis in human colorectal cancer cells. (A) HCT116 and SW480 cells were treated with INDO (300 μM) or SUS (125 μM for HCT116 and 200 μM for SW480 cells). Cells were collected at the indicated times and scored for morphological evidence of apoptosis as described in Materials and Methods. (B) HCT116 and SW480 cells were treated with SUS or INDO for 60 hr, harvested, and assessed for morphological signs of apoptosis. Cells treated with SUS or INDO show the hallmarks of apoptosis. Independent evidence of apoptosis was obtained by examining phosphatidylserine exposure. SUS treatment of HCT116 and SW480 cells resulted in substantial phosphatidylserine exposure indicative of membrane unpacking and apoptosis. Forty-eight hours after treatment with 125 μM SUS, 44% of HCT116 were apoptotic as judged by phosphatidylserine exposure vs. 4% in the vehicle-treated control cells. Likewise, 74% of SW480 cells were apoptotic 50 hr after treatment with 200 μM SUS vs. 1.5% of the vehicle-treated control cells. (C) CHX inhibits SUS-induced apoptosis in HCT116 and SW480 cells. Cells were treated with SUS as above in the presence or absence of CHX (10 μM). Apoptotic cells were evaluated as described above.
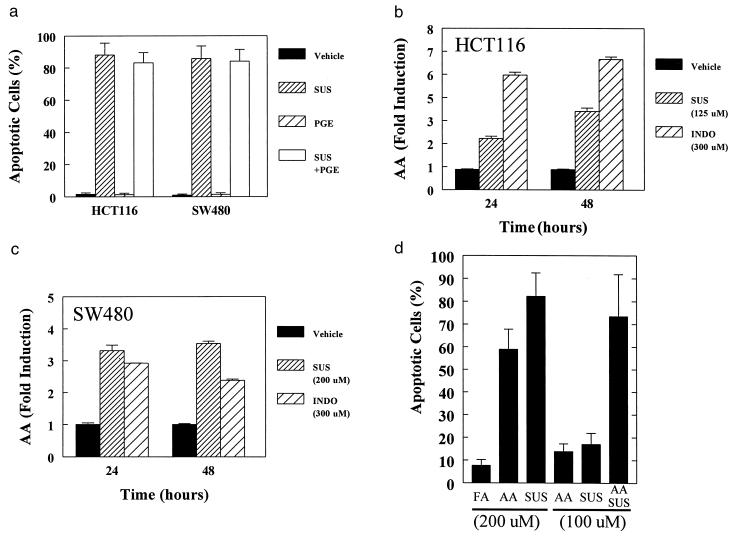
AA mediates NSAID-induced apoptosis. (A) Prostaglandin E2 does not inhibit SUS-induced apoptosis. Cells were treated with the indicated combination of SUS (same concentrations as in Fig. 1) and prostaglandin E2 (PGE, 10 μM). (B, C) Increased AA levels after NSAID treatment. HCT116 and SW480 cells were treated with the indicated concentrations of SUS or INDO. AA levels were determined by measuring release of 3H-AA into the media as described in the Methods. Data are indicated as the mean of three replicates. Error bars = SD. Independent confirmation of AA increases were obtained by gas chromatographic evaluation of AA levels. Treatment of HCT116 and SW480 cells with SUS for 16 hr as described in Fig. 1 resulted in a 2.3- and 3.1-fold increase in AA levels. (D) AA can mimic the effects of SUS treatment and act synergistically with SUS to induce apoptosis. SW480 cells were treated with AA, SUS, or behenic acid as a control fatty acid (FA) as indicated. After 48 hr of treatment, apoptosis was assessed and expressed as percent apoptotic cells − percent apoptotic cells in untreated cells (5.6% ± 2.4). Data are indicated as the mean of duplicates. Error bars = SD.
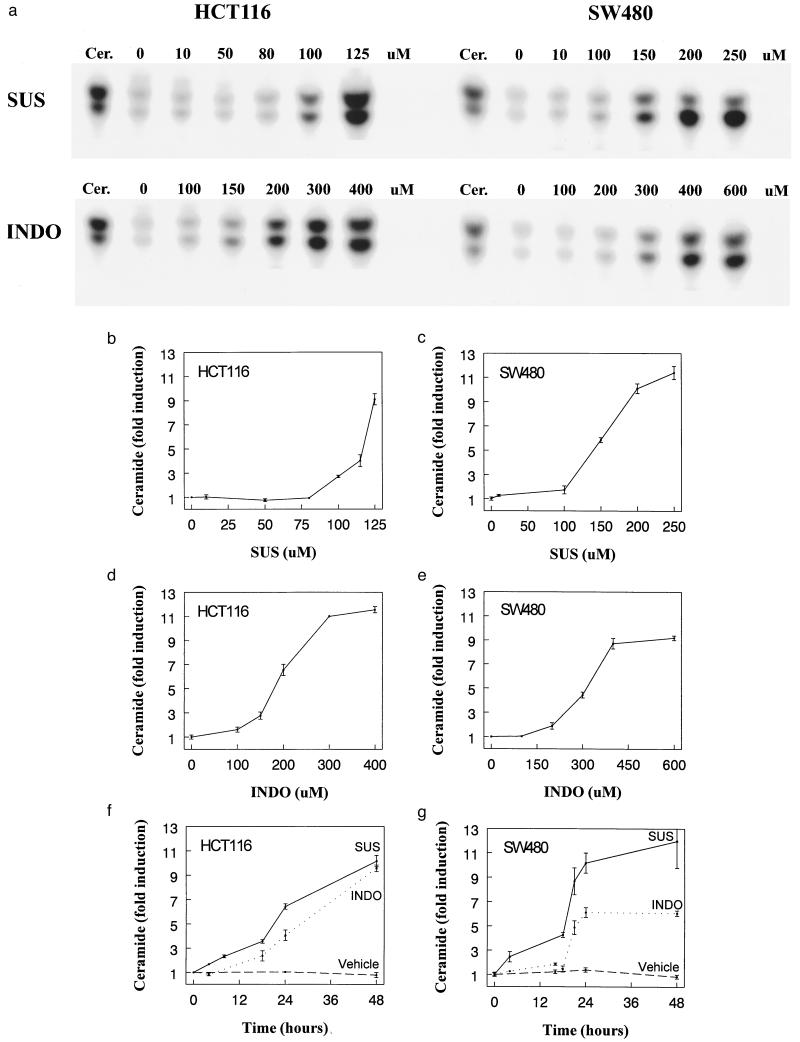
NSAID treatment induces ceramide generation. (A) Ceramide increases in a dose-dependent fashion in colon cancer cells treated with SUS or INDO. HCT116 and SW480 cells were treated with the indicated concentration of SUS and INDO for 48 hr. Cells were harvested and ceramide was quantified as described in Materials and Methods. The first lane of each panel corresponds to a ceramide standard (ceramide type III, Sigma). (B–E) Dose response of ceramide induction. HCT116 and SW480 cells were treated with SUS or INDO as indicated. Ceramide levels were determined as described in the Methods. Data shows mean of at least two experiments. Error bars = SD. Dimethyl sulfoxide vehicle controls (using equivalent volumes) were performed in parallel with the drug treatments; no significant ceramide induction resulted (not shown). (F, G) Time course of ceramide induction. HCT116 and SW480 cells were treated with SUS (125 μM for HCT116 and 200 μM for SW480) or INDO (300 μM). Control cells were treated with dimethyl sulfoxide vehicle only. Cells were harvested at the indicated times following treatment and ceramide levels were determined as described in Materials and Methods. The data are plotted as the mean of at least two experiments. Error bars = SD.
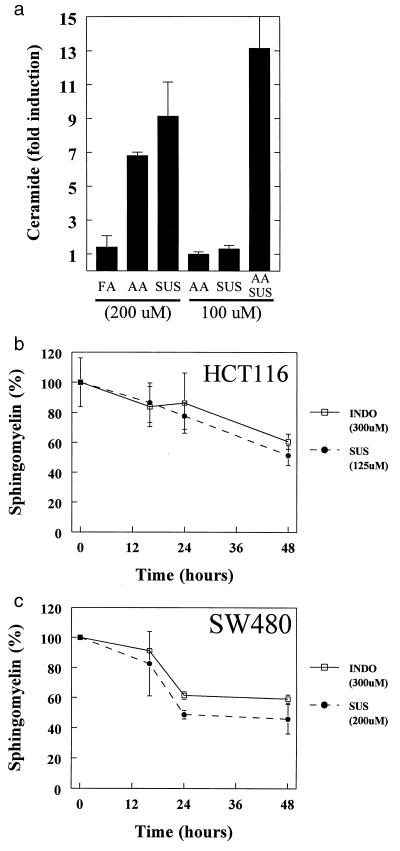
Ceramide production in NSAID-mediated apoptosis. (A) AA can mimic the effects of SUS treatment and act synergistically with SUS to induce ceramide. SW480 cells were treated with AA and SUS as indicated. After 48 hr of treatment ceramide increases were assessed and are expressed as fold increases relative to untreated cells. Data are indicated as the mean of duplicates. Error bars = SD. (B, C) Induction of sphingomyelin turnover in response to NSAID treatment. Sphingomyelin levels at each time point are plotted as a percentage of that in untreated control cells. For each point, the mean of at least two independent experiments is presented. Error bars = SD.
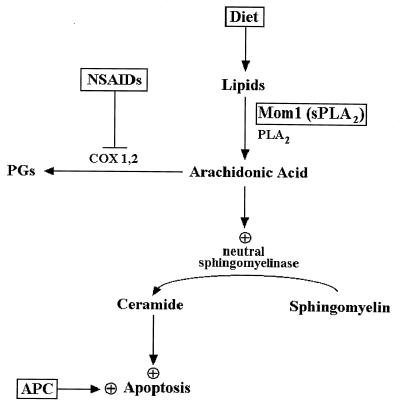
Model relating AA and ceramide to colorectal cancer chemoprevention. Mutation of the APC gene initiates colorectal tumorigenesis and results in abnormally decreased apoptosis (16). Perturbation of lipid metabolism by pharmacological, dietary, or genetic means can partially correct the deficits. AA is generated by cytosolic and secreted phospholipase A2, which hydrolyzes plasma membrane lipids or lipids derived from the diet. AA is normally used as a substrate by the COXs to produce eicosanoids such as prostaglandins. NSAIDs inhibit the activity of the COXs, which increases the cellular pool of AA. AA stimulates neutral sphingomyelinase activity (18, 37), which catalyzes the hydrolysis of sphingomyelin to generate ceramide. Ceramide acts as second messenger that activates the cellular apoptotic machinery. The Mom1 gene is a modifier of polyp formation in the Min mouse, a model for APC mutation-induced colon tumors. The Mom1 gene encodes a secreted phospholipase A2, an enzyme predicted to increase the level of free AA. Inactivating mutations in Mom1 predispose Min mice to developing intestinal polyps and would be expected to result in reduced levels of AA (–36). This in turn could result in decreased production of ceramide and therefore a relative resistance to programmed cell death and increased tumor susceptibility. The lipid compositions of diets are known to affect colon cancer risk (41, 42). Diets rich in unsaturated fatty acids such as AA are associated with a decreased incidence of colon cancer (43). This effect could be due to increased levels of AA and subsequent increased susceptibility to apoptosis as described above.
Similar articles
-
COX-2, NSAIDs and human neoplasia. Part I: Colorectal neoplasms.In Vivo. 2002 Nov-Dec;16(6):501-9. In Vivo. 2002. PMID: 12494894 Review.
-
The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells.Proc Natl Acad Sci U S A. 2003 Aug 19;100(17):9968-73. doi: 10.1073/pnas.1631086100. Epub 2003 Aug 8. Proc Natl Acad Sci U S A. 2003. PMID: 12909723 Free PMC article.
-
Cyclooxygenase expression is not required for release of arachidonic acid from cells by some nonsteroidal anti-inflammatory drugs and cancer preventive agents.BMC Pharmacol. 2006 Mar 29;6:7. doi: 10.1186/1471-2210-6-7. BMC Pharmacol. 2006. PMID: 16571133 Free PMC article.
-
The role of arachidonic acid regulatory enzymes in colorectal disease.Dis Colon Rectum. 2005 Jul;48(7):1471-83. doi: 10.1007/s10350-005-0015-y. Dis Colon Rectum. 2005. PMID: 15868226 Review.
-
Nonsteroidal anti-inflammatory drugs and oxidative stress in cancer cells.Histol Histopathol. 2007 Apr;22(4):437-42. doi: 10.14670/HH-22.437. Histol Histopathol. 2007. PMID: 17290354 Review.
Cited by
-
The expression of phospholipase A2 group X is inversely associated with metastasis in colorectal cancer.Oncol Lett. 2013 Feb;5(2):533-538. doi: 10.3892/ol.2012.1067. Epub 2012 Dec 10. Oncol Lett. 2013. PMID: 23420493 Free PMC article.
-
Systems Pharmacology Dissection of the Integrated Treatment for Cardiovascular and Gastrointestinal Disorders by Traditional Chinese Medicine.Sci Rep. 2016 Sep 6;6:32400. doi: 10.1038/srep32400. Sci Rep. 2016. PMID: 27597117 Free PMC article.
-
Impact of group IVA cytosolic phospholipase A2 gene polymorphisms on phenotypic features of patients with familial adenomatous polyposis.Int J Colorectal Dis. 2010 Mar;25(3):293-301. doi: 10.1007/s00384-009-0808-x. Epub 2009 Oct 1. Int J Colorectal Dis. 2010. PMID: 19795129
-
Ion channel inhibitors block caspase activation by mechanisms other than restoring intracellular potassium concentration.Cell Death Dis. 2011 Jan 13;2(1):e113. doi: 10.1038/cddis.2010.93. Cell Death Dis. 2011. PMID: 21368885 Free PMC article.
-
Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits apoptosis.Mol Cell Biol. 2001 Sep;21(18):6322-31. doi: 10.1128/MCB.21.18.6322-6331.2001. Mol Cell Biol. 2001. PMID: 11509673 Free PMC article.
References
-
- Giardiello F M. Gastroenterol Clin North Am. 1996;25:349–362. - PubMed
-
- Beazer-Barclay Y, Levy D B, Moser A M, Dove W F, Hamilton S R, Vogelstein B, Kinzler K W. Carcinogenesis. 1996;17:1757–1760. - PubMed
-
- Boolbol S K, Dannenberg A J, Chadurn A, Martucci C, Guo X, Ramonetti J T, Abreu-Goris M, Newmark H L, Lipkin M L, DeCosse J J, Bertagnolli M M. Cancer Res. 1996;56:2556–2560. - PubMed
-
- Jacoby R F, Marshall D J, Newton M A, Novakovic K, Tutsch K, Cole C E, Lubet R A, Kelloff G J, Verma A, Moser A R, Dove W F. Cancer Res. 1996;56:710–714. - PubMed
-
- Thun M J. Gastroenterol Clin North Am. 1996;25:333–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
