

Biallelic variants in the calpain regulatory subunit CAPNS1 cause pulmonary arterial hypertension
- PMID: 38230350
- PMCID: PMC10790724
- DOI: 10.1016/j.gimo.2023.100811
Biallelic variants in the calpain regulatory subunit CAPNS1 cause pulmonary arterial hypertension
Abstract
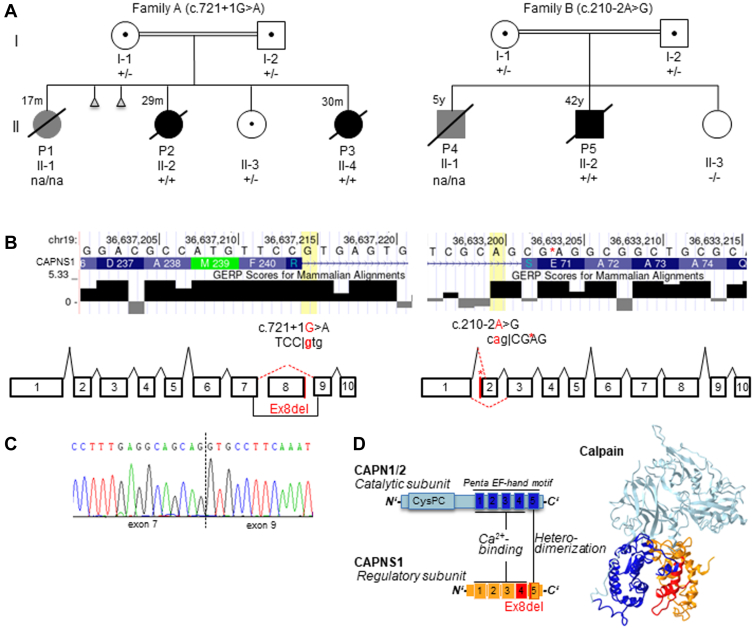
Purpose: The aim of this study was to identify the monogenic cause of pulmonary arterial hypertension (PAH), a multifactorial and often fatal disease, in 2 unrelated consanguine families.
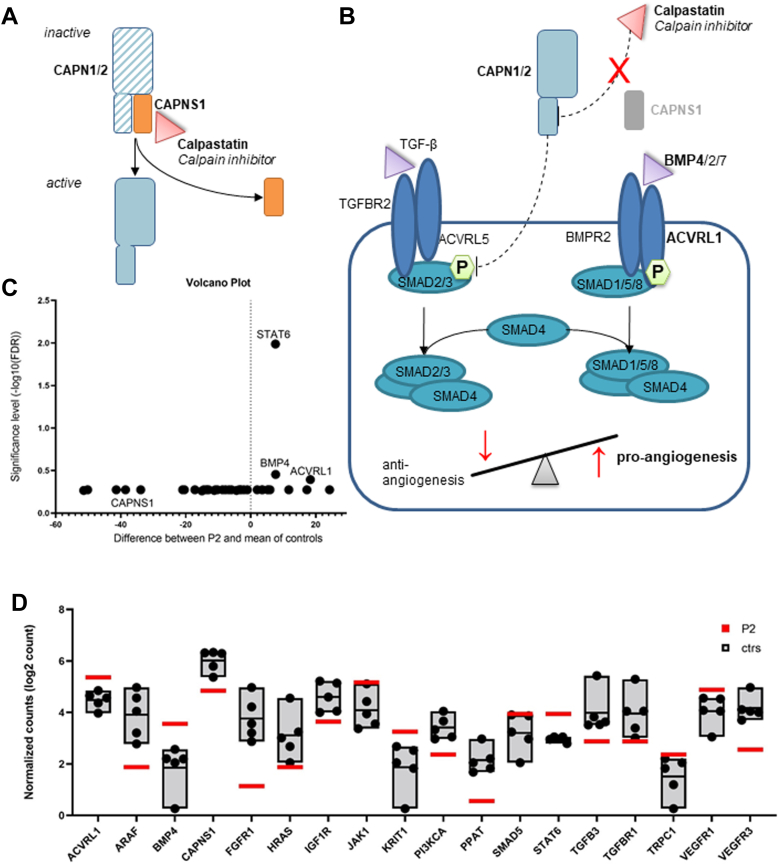
Methods: We performed exome sequencing and validated variant pathogenicity by whole-blood RNA and protein expression analysis in both families. Further RNA sequencing of preserved lung tissue was performed to investigate the consequences on selected genes that are involved in angiogenesis, proliferation, and apoptosis.
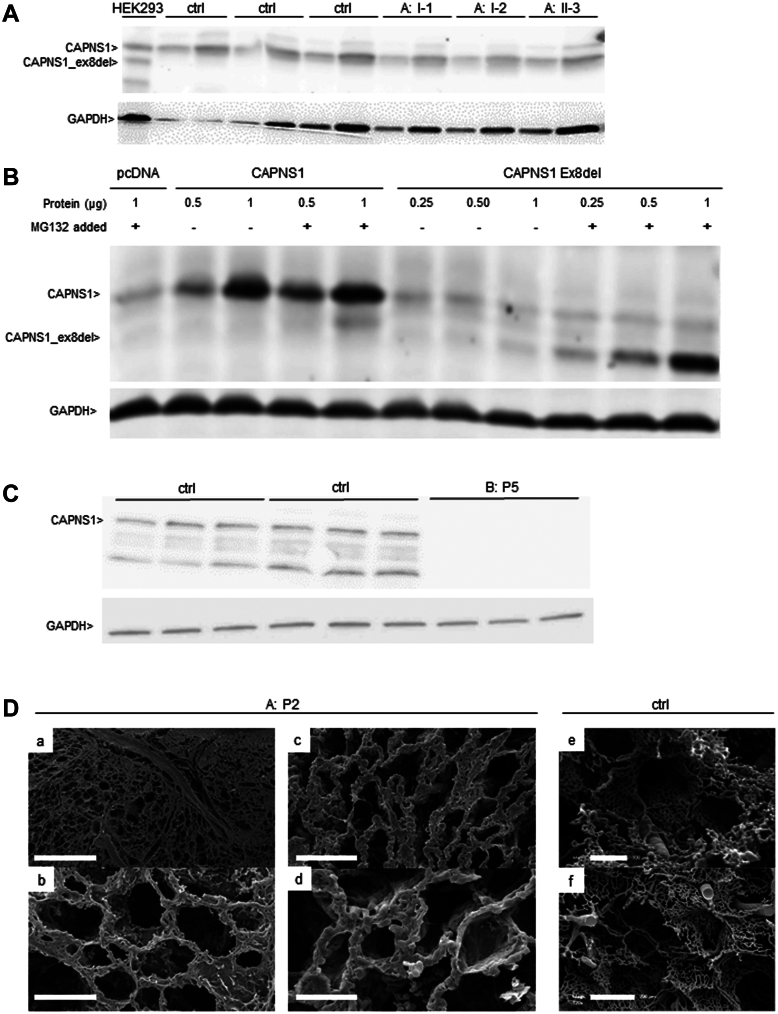
Results: We identified 2 rare biallelic variants in CAPNS1, encoding the regulatory subunit of calpain. The variants cosegregated with PAH in the families. Both variants lead to loss of function (LoF), which is demonstrated by aberrant splicing resulting in the complete absence of the CAPNS1 protein in affected patients. No other LoF CAPNS1 variant was identified in the genome data of more than 1000 patients with unresolved PAH.
Conclusion: The calpain holoenzyme was previously linked to pulmonary vascular development and progression of PAH in patients. We demonstrated that biallelic LoF variants in CAPNS1 can cause idiopathic PAH by the complete absence of CAPNS1 protein. Screening of this gene in patients who are affected by PAH, especially with suspected autosomal recessive inheritance, should be considered.
Keywords: Angiogenesis; CAPNS1; Calpain; Pulmonary arterial hypertension.
© 2023 The Authors.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Rare variant analysis of 4241 pulmonary arterial hypertension cases from an international consortium implicates FBLN2, PDGFD, and rare de novo variants in PAH.Genome Med. 2021 May 10;13(1):80. doi: 10.1186/s13073-021-00891-1. Genome Med. 2021. PMID: 33971972 Free PMC article.
-
Phenotypic Characterization of EIF2AK4 Mutation Carriers in a Large Cohort of Patients Diagnosed Clinically With Pulmonary Arterial Hypertension.Circulation. 2017 Nov 21;136(21):2022-2033. doi: 10.1161/CIRCULATIONAHA.117.028351. Epub 2017 Sep 28. Circulation. 2017. PMID: 28972005 Free PMC article.
-
Biallelic variants of ATP13A3 cause dose-dependent childhood-onset pulmonary arterial hypertension characterised by extreme morbidity and mortality.J Med Genet. 2022 Sep;59(9):906-911. doi: 10.1136/jmedgenet-2021-107831. Epub 2021 Sep 7. J Med Genet. 2022. PMID: 34493544 Free PMC article.
-
The calpain system as a modulator of stress/damage response.Cell Cycle. 2007 Jan 15;6(2):136-8. doi: 10.4161/cc.6.2.3759. Epub 2007 Jan 27. Cell Cycle. 2007. PMID: 17264674 Review.
-
Description of Two New Cases of AQP1 Related Pulmonary Arterial Hypertension and Review of the Literature.Genes (Basel). 2022 May 22;13(5):927. doi: 10.3390/genes13050927. Genes (Basel). 2022. PMID: 35627312 Free PMC article. Review.
References
-
- Tuder R.M., Ponticos M., Holmes A. In: Scleroderma: From Pathogenesis to Comprehensive Management. Varga J., Denton C., Wigley F., Allanore Y., Kuwana M., editors. Springer International Publishing; 2017. Pathogenesis of pulmonary arterial hypertension; pp. 385–401.
LinkOut - more resources
Full Text Sources
Miscellaneous
