

Interplay between mitochondrial reactive oxygen species, oxidative stress and hypoxic adaptation in facioscapulohumeral muscular dystrophy: Metabolic stress as potential therapeutic target
- PMID: 35248827
- PMCID: PMC8899416
- DOI: 10.1016/j.redox.2022.102251
Interplay between mitochondrial reactive oxygen species, oxidative stress and hypoxic adaptation in facioscapulohumeral muscular dystrophy: Metabolic stress as potential therapeutic target
Abstract
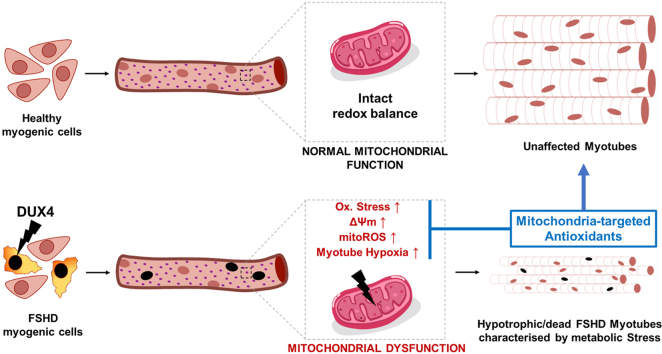
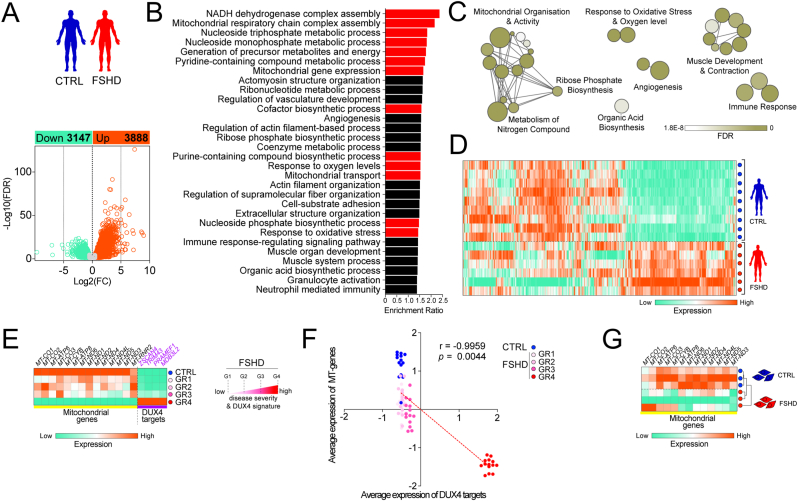
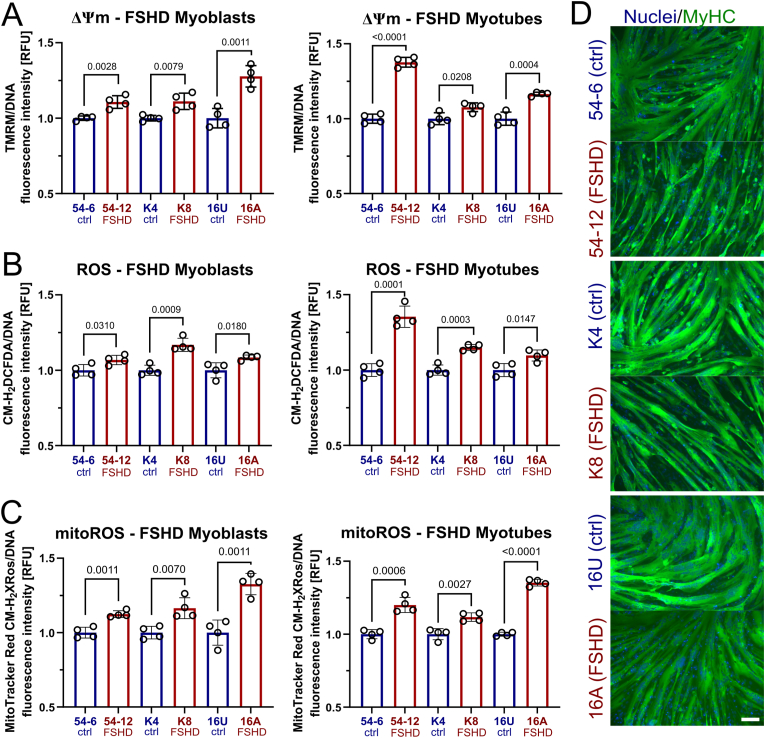
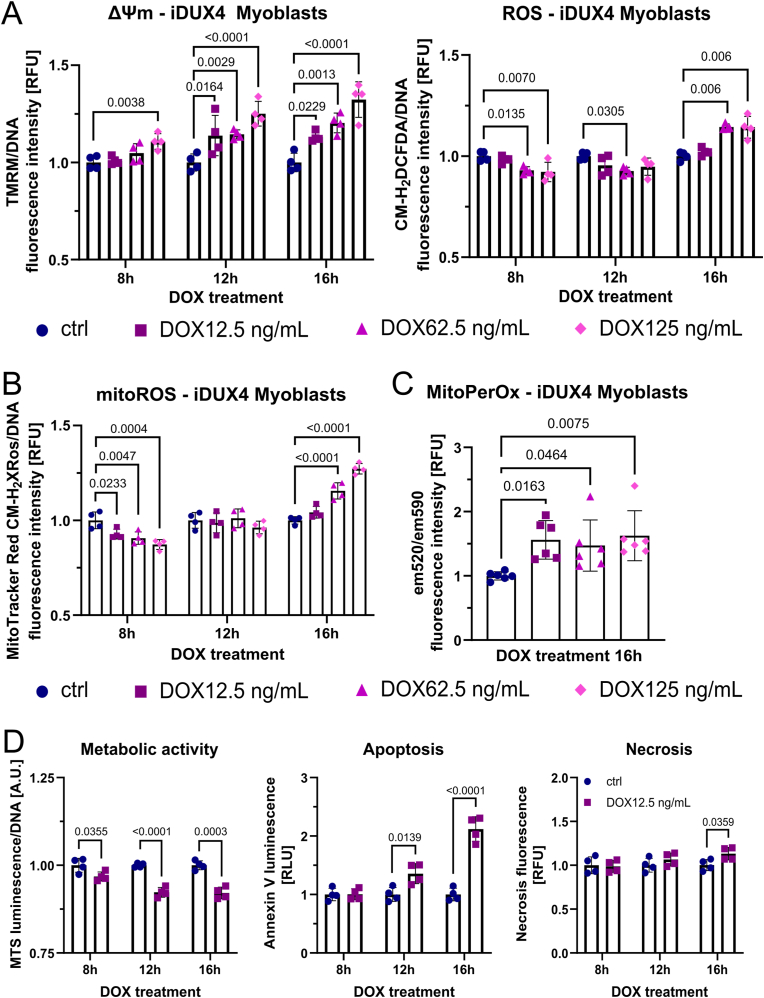
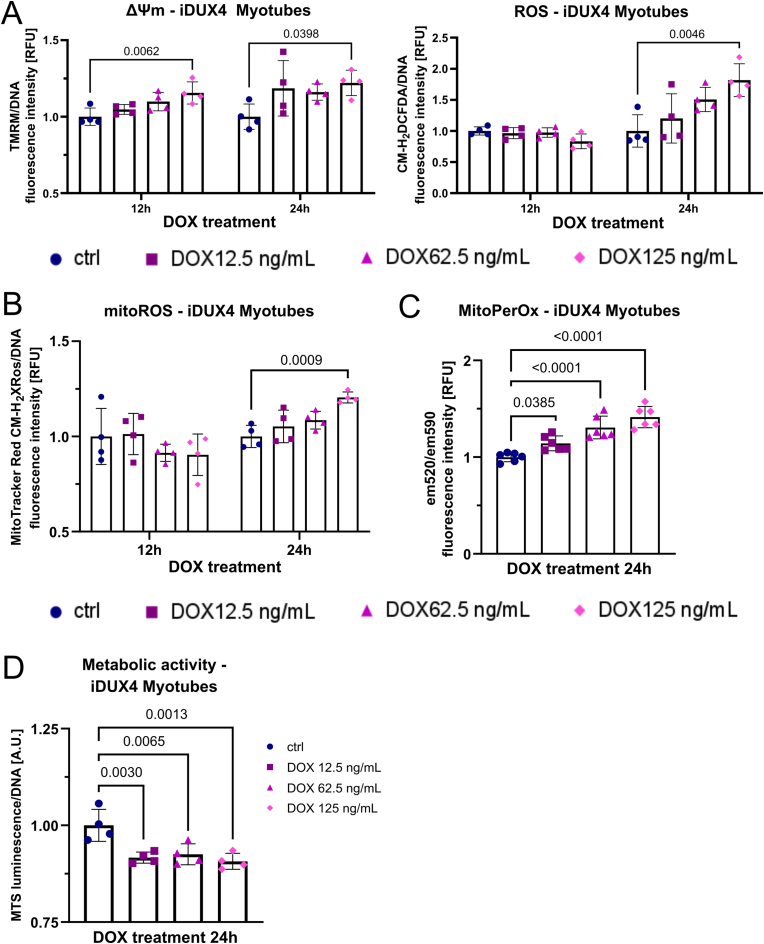
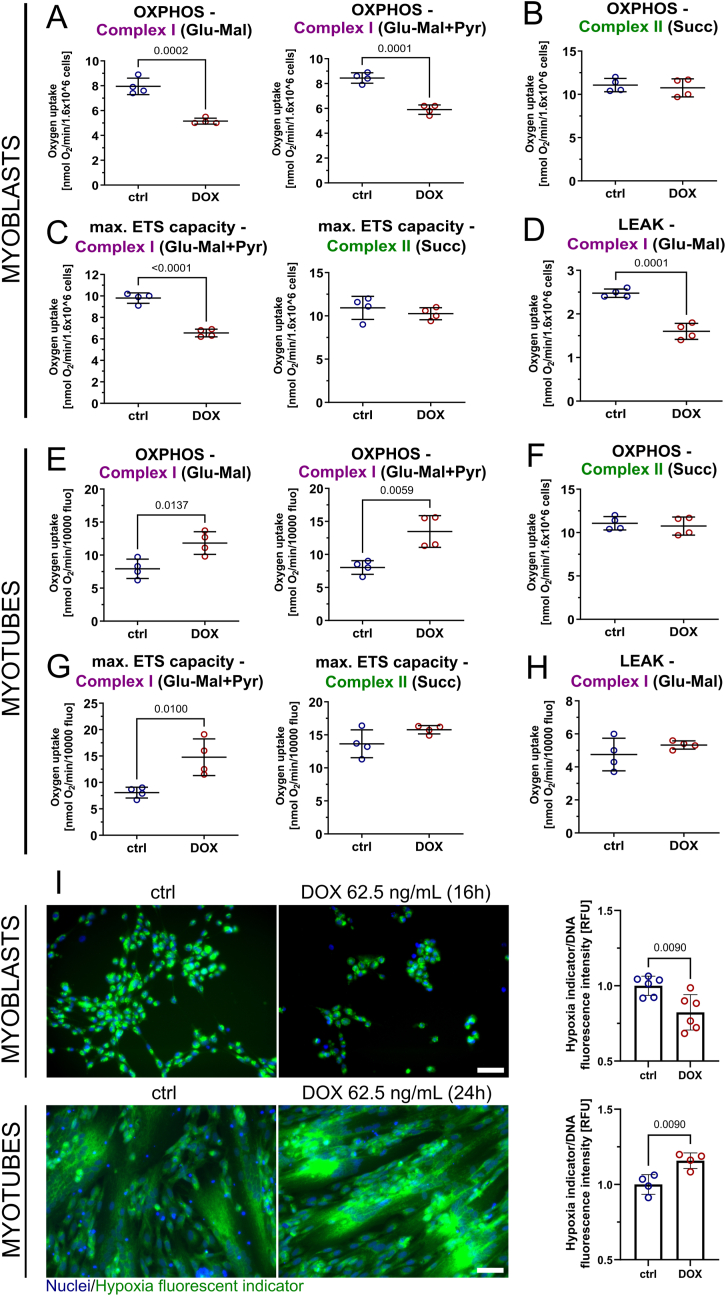
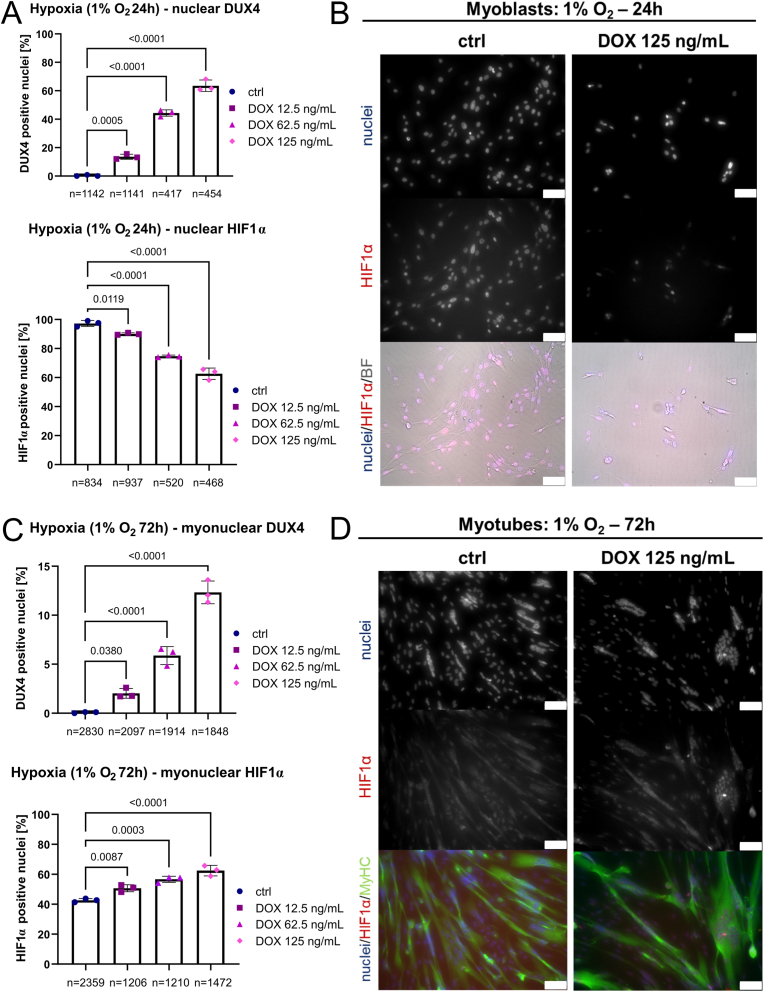
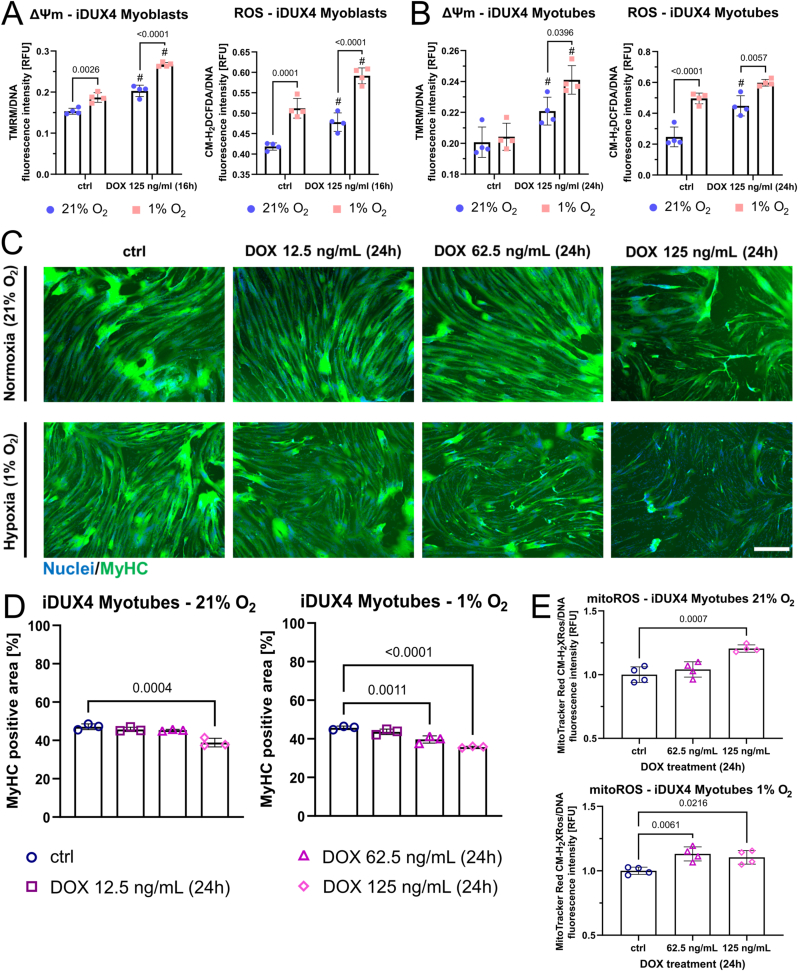
Facioscapulohumeral muscular dystrophy (FSHD) is characterised by descending skeletal muscle weakness and wasting. FSHD is caused by mis-expression of the transcription factor DUX4, which is linked to oxidative stress, a condition especially detrimental to skeletal muscle with its high metabolic activity and energy demands. Oxidative damage characterises FSHD and recent work suggests metabolic dysfunction and perturbed hypoxia signalling as novel pathomechanisms. However, redox biology of FSHD remains poorly understood, and integrating the complex dynamics of DUX4-induced metabolic changes is lacking. Here we pinpoint the kinetic involvement of altered mitochondrial ROS metabolism and impaired mitochondrial function in aetiology of oxidative stress in FSHD. Transcriptomic analysis in FSHD muscle biopsies reveals strong enrichment for pathways involved in mitochondrial complex I assembly, nitrogen metabolism, oxidative stress response and hypoxia signalling. We found elevated mitochondrial ROS (mitoROS) levels correlate with increases in steady-state mitochondrial membrane potential in FSHD myogenic cells. DUX4 triggers mitochondrial membrane polarisation prior to oxidative stress generation and apoptosis through mitoROS, and affects mitochondrial health through lipid peroxidation. We identify complex I as the primary target for DUX4-induced mitochondrial dysfunction, with strong correlation between complex I-linked respiration and cellular oxygenation/hypoxia signalling activity in environmental hypoxia. Thus, FSHD myogenesis is uniquely susceptible to hypoxia-induced oxidative stress as a consequence of metabolic mis-adaptation. Importantly, mitochondria-targeted antioxidants rescue FSHD pathology more effectively than conventional antioxidants, highlighting the central involvement of disturbed mitochondrial ROS metabolism. This work provides a pathomechanistic model by which DUX4-induced changes in oxidative metabolism impair muscle function in FSHD, amplified when metabolic adaptation to varying O2 tension is required.
Keywords: Antioxidants; DUX4; Facioscapulohumeral muscular dystrophy; Hypoxia; Mitochondrial dysfunction; Reactive oxygen species.
Copyright © 2022. Published by Elsevier B.V.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
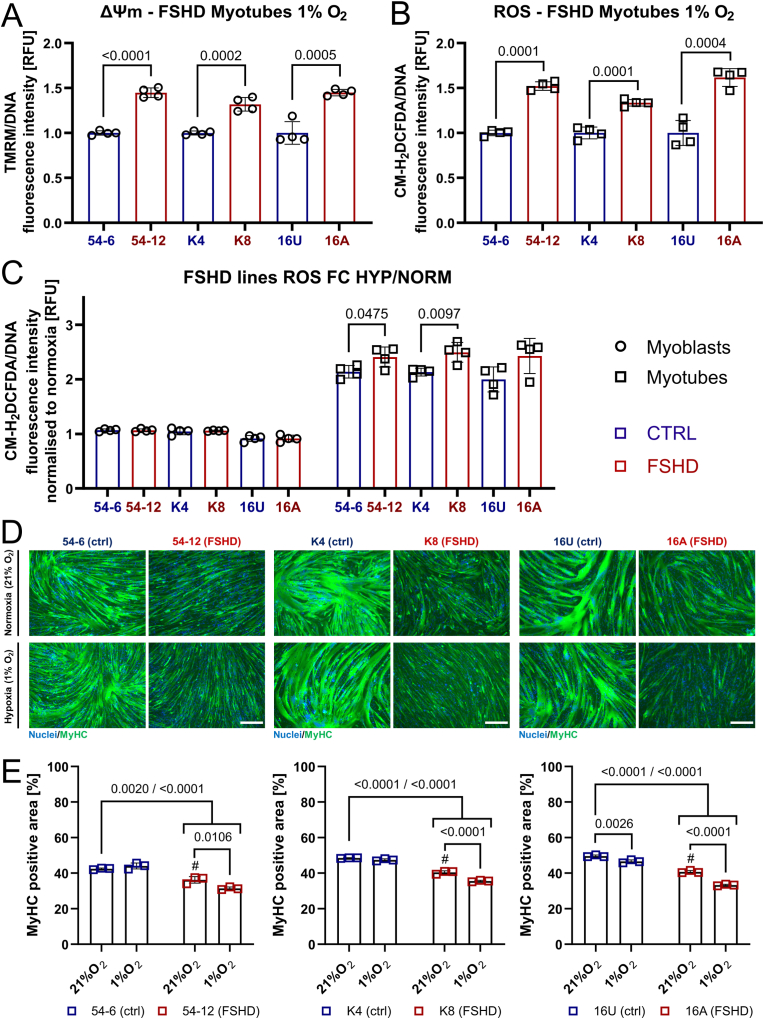
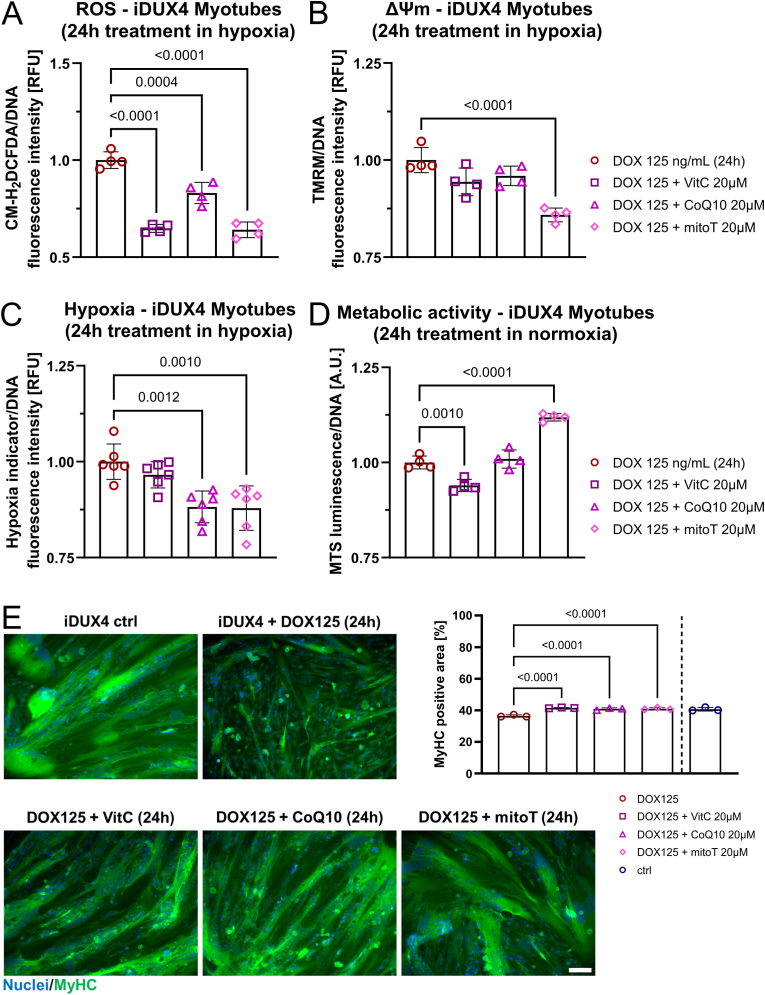
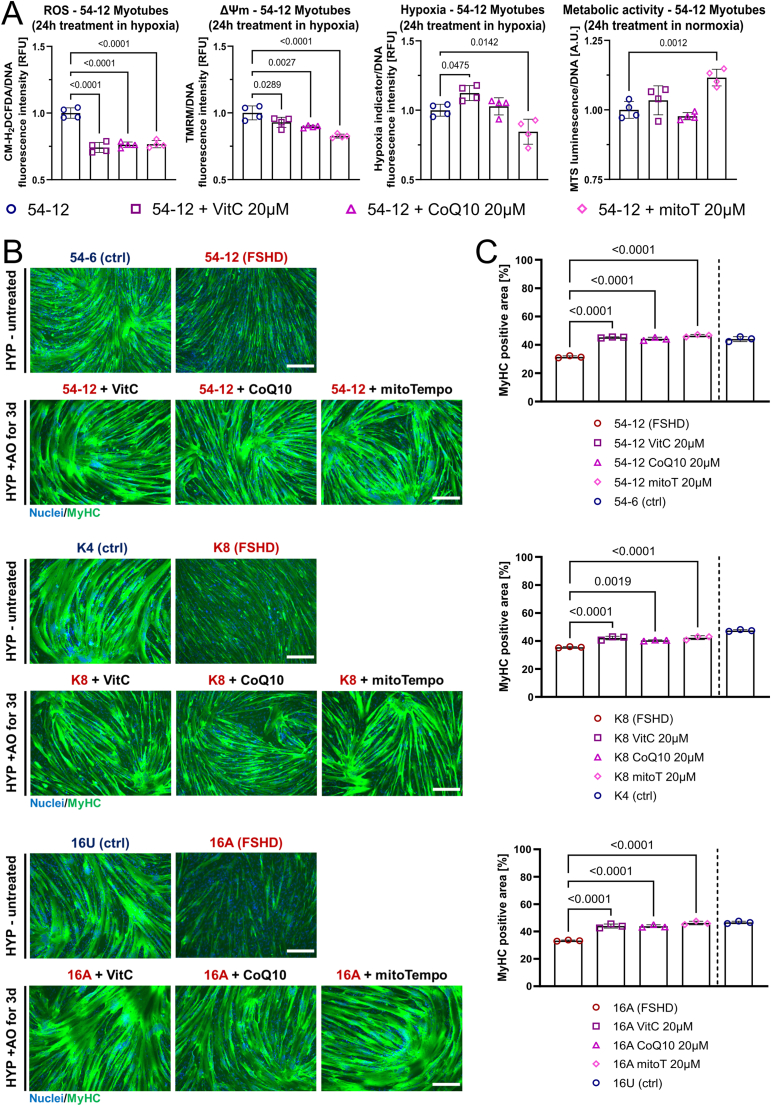
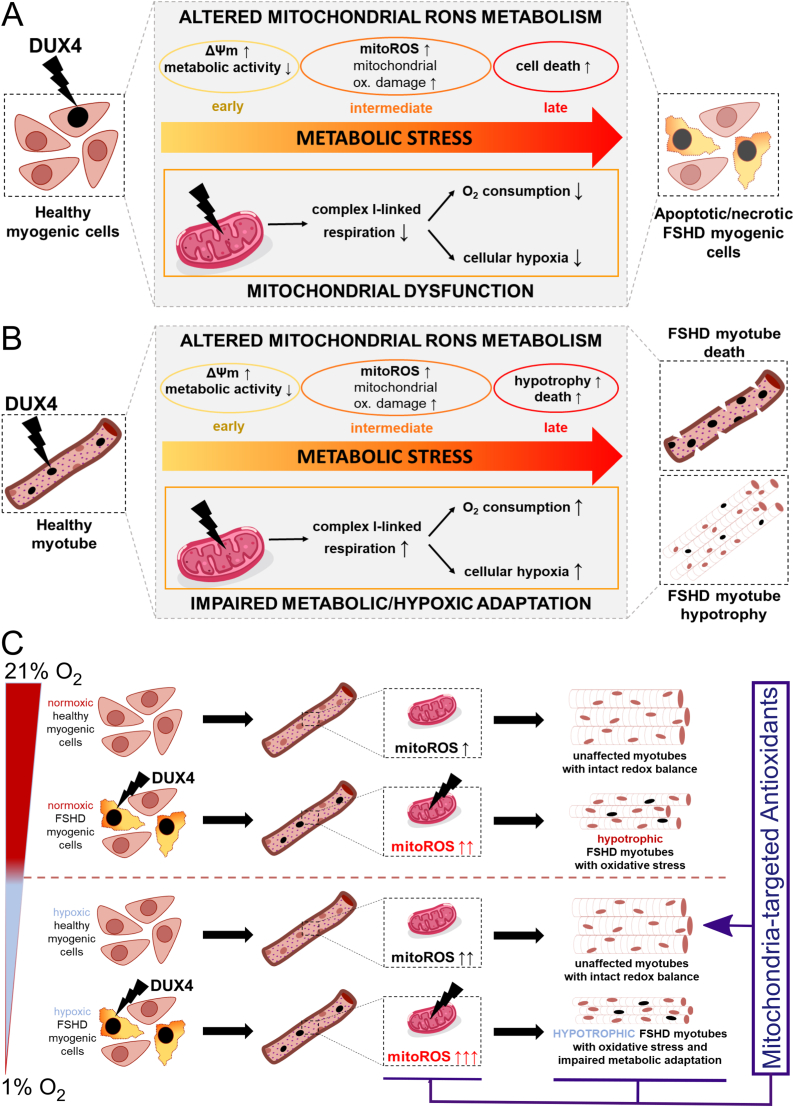
Similar articles
-
SIX transcription factors are necessary for the activation of DUX4 expression in facioscapulohumeral muscular dystrophy.Skelet Muscle. 2024 Dec 3;14(1):30. doi: 10.1186/s13395-024-00361-3. Skelet Muscle. 2024. PMID: 39627769 Free PMC article.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Muscle Proteome Analysis of Facioscapulohumeral Dystrophy Patients Reveals a Metabolic Rewiring Promoting Oxidative/Reductive Stress Contributing to the Loss of Muscle Function.Antioxidants (Basel). 2024 Nov 16;13(11):1406. doi: 10.3390/antiox13111406. Antioxidants (Basel). 2024. PMID: 39594549 Free PMC article.
-
The DUX4-HIF1α Axis in Murine and Human Muscle Cells: A Link More Complex Than Expected.Int J Mol Sci. 2024 Mar 15;25(6):3327. doi: 10.3390/ijms25063327. Int J Mol Sci. 2024. PMID: 38542301 Free PMC article.
-
Undernutrition as a risk factor for tuberculosis disease.Cochrane Database Syst Rev. 2024 Jun 11;6(6):CD015890. doi: 10.1002/14651858.CD015890.pub2. Cochrane Database Syst Rev. 2024. PMID: 38860538 Free PMC article. Review.
Cited by
-
The transcription factor DUX4 orchestrates translational reprogramming by broadly suppressing translation efficiency and promoting expression of DUX4-induced mRNAs.PLoS Biol. 2023 Sep 25;21(9):e3002317. doi: 10.1371/journal.pbio.3002317. eCollection 2023 Sep. PLoS Biol. 2023. PMID: 37747887 Free PMC article.
-
Modulation of neural circuits by melatonin in neurodegenerative and neuropsychiatric disorders.Naunyn Schmiedebergs Arch Pharmacol. 2024 Jun;397(6):3867-3895. doi: 10.1007/s00210-023-02939-y. Epub 2024 Jan 16. Naunyn Schmiedebergs Arch Pharmacol. 2024. PMID: 38225412 Review.
-
The Role of Oxidative Stress-Mediated Inflammation in the Development of T2DM-Induced Diabetic Nephropathy: Possible Preventive Action of Tannins and Other Oligomeric Polyphenols.Molecules. 2022 Dec 18;27(24):9035. doi: 10.3390/molecules27249035. Molecules. 2022. PMID: 36558167 Free PMC article. Review.
-
The FSHD muscle-blood biomarker: a circulating transcriptomic biomarker for clinical severity in facioscapulohumeral muscular dystrophy.Brain Commun. 2023 Aug 16;5(5):fcad221. doi: 10.1093/braincomms/fcad221. eCollection 2023. Brain Commun. 2023. PMID: 37731904 Free PMC article.
-
Flavones provide resistance to DUX4-induced toxicity via an mTor-independent mechanism.Cell Death Dis. 2023 Nov 16;14(11):749. doi: 10.1038/s41419-023-06257-2. Cell Death Dis. 2023. PMID: 37973788 Free PMC article.
References
-
- Sposito R., et al. Facioscapulohumeral muscular dystrophy type 1A in northwestern Tuscany: a molecular genetics-based epidemiological and genotype-phenotype study. Genet. Test. 2005;9(1):30–36. - PubMed
-
- Tawil R., Van Der Maarel S.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34(1):1–15. - PubMed
-
- Pandya S., King W.M., Tawil R. Facioscapulohumeral dystrophy. Phys. Ther. 2008;88(1):105–113. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
