

Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer's disease-like neuroinflammation and cognitive impairment
- PMID: 34526055
- PMCID: PMC8442347
- DOI: 10.1186/s13024-021-00488-7
Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer's disease-like neuroinflammation and cognitive impairment
Abstract
Background: Human genetic association studies point to immune response and lipid metabolism, in addition to amyloid-beta (Aβ) and tau, as major pathways in Alzheimer's disease (AD) etiology. Accumulating evidence suggests that chronic neuroinflammation, mainly mediated by microglia and astrocytes, plays a causative role in neurodegeneration in AD. Our group and others have reported early and dramatic losses of brain sulfatide in AD cases and animal models that are mediated by ApoE in an isoform-dependent manner and accelerated by Aβ accumulation. To date, it remains unclear if changes in specific brain lipids are sufficient to drive AD-related pathology.
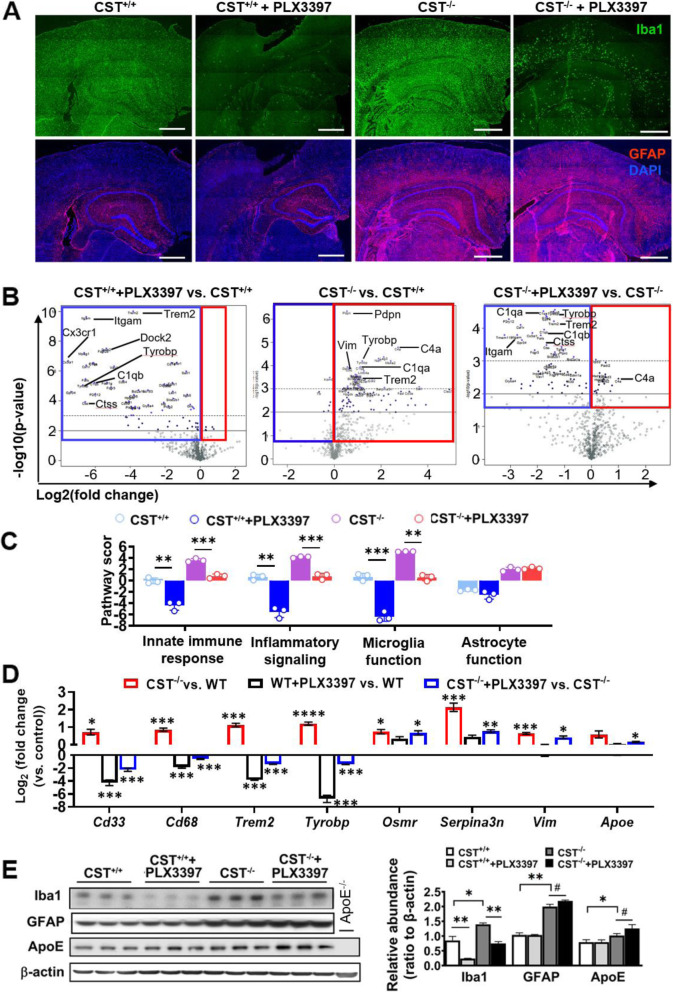
Methods: To study the consequences of CNS sulfatide deficiency and gain insights into the underlying mechanisms, we developed a novel mouse model of adult-onset myelin sulfatide deficiency, i.e., tamoxifen-inducible myelinating glia-specific cerebroside sulfotransferase (CST) conditional knockout mice (CSTfl/fl/Plp1-CreERT), took advantage of constitutive CST knockout mice (CST-/-), and generated CST/ApoE double knockout mice (CST-/-/ApoE-/-), and assessed these mice using a broad range of methodologies including lipidomics, RNA profiling, behavioral testing, PLX3397-mediated microglia depletion, mass spectrometry (MS) imaging, immunofluorescence, electron microscopy, and Western blot.
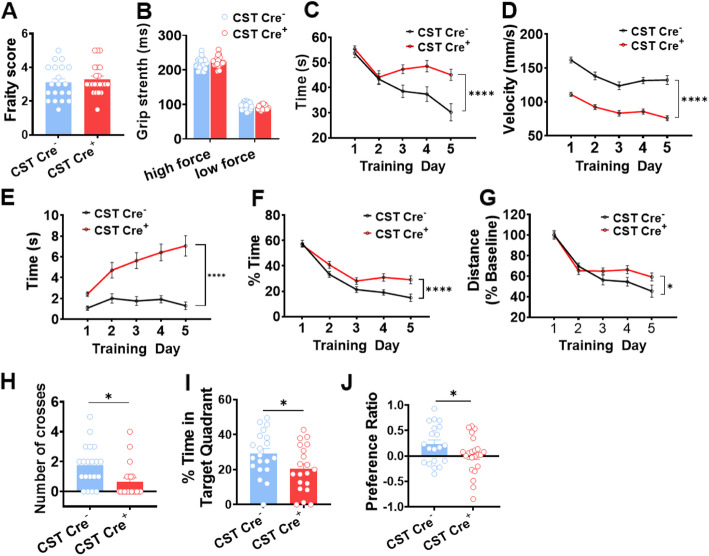
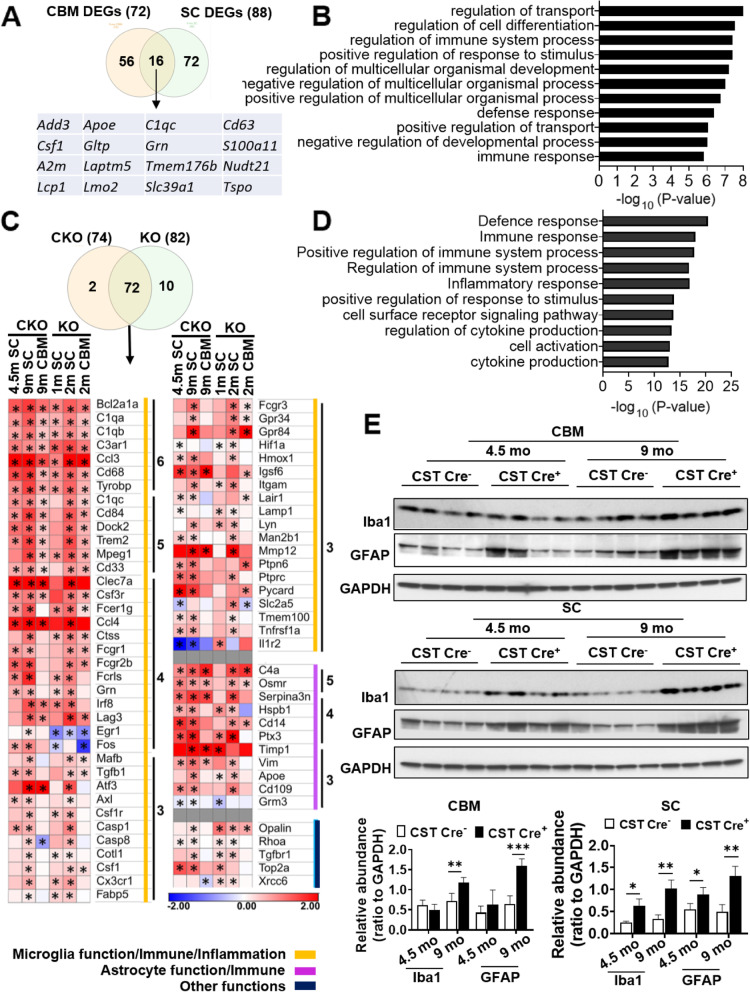
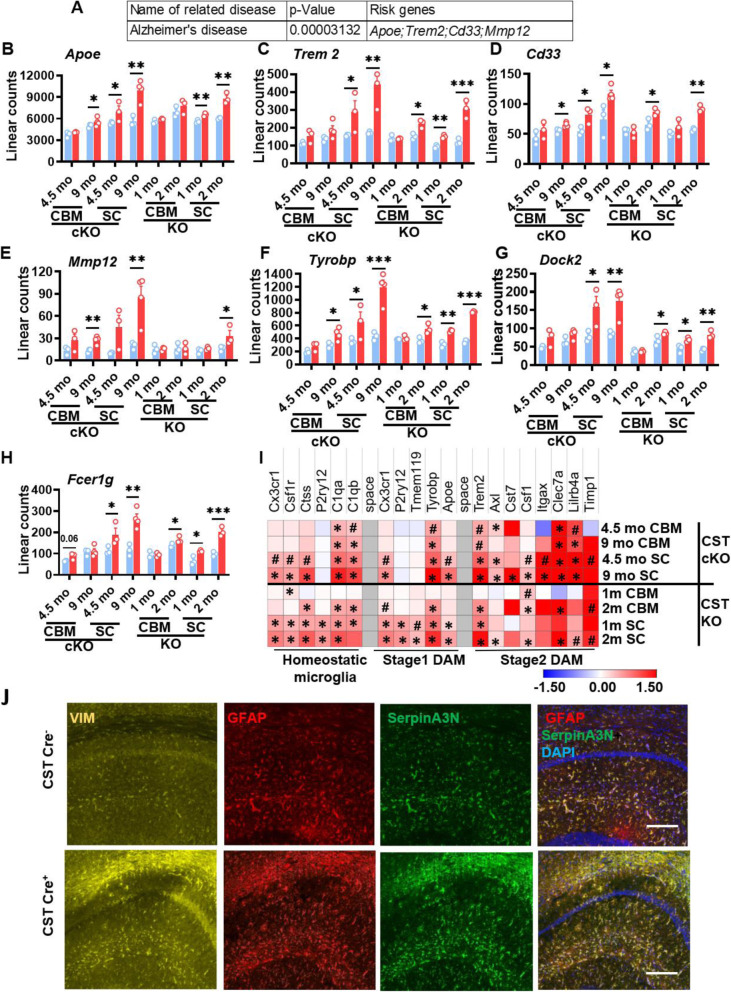
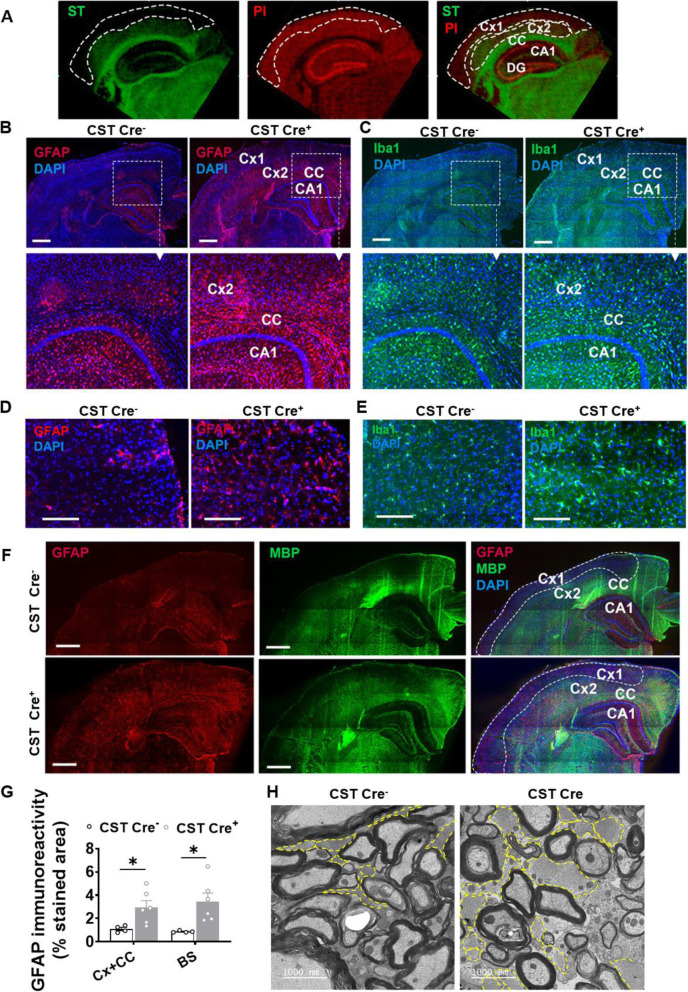
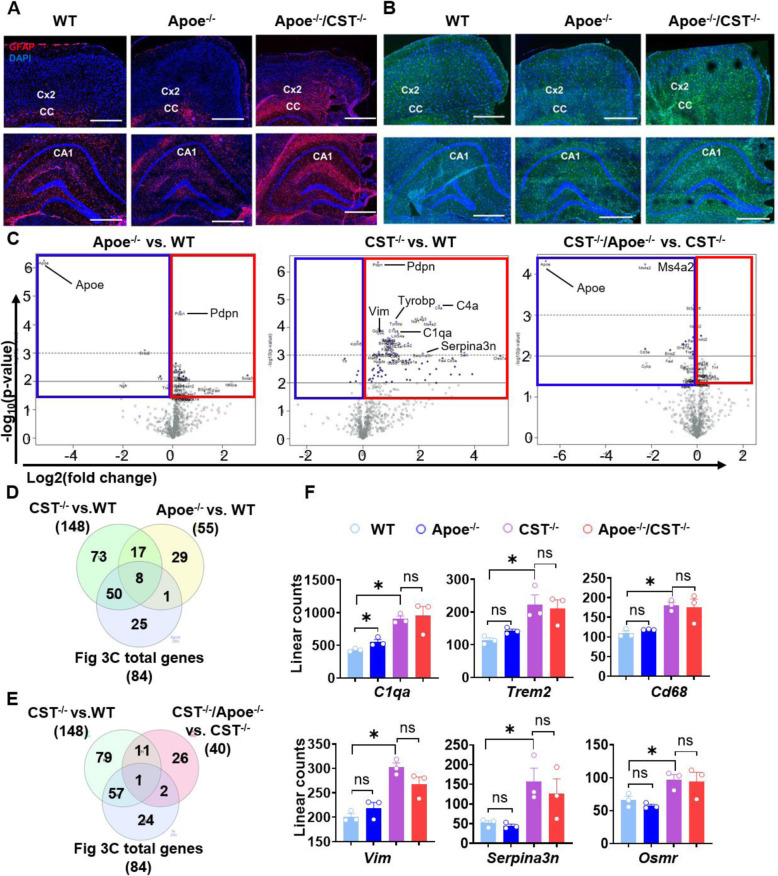
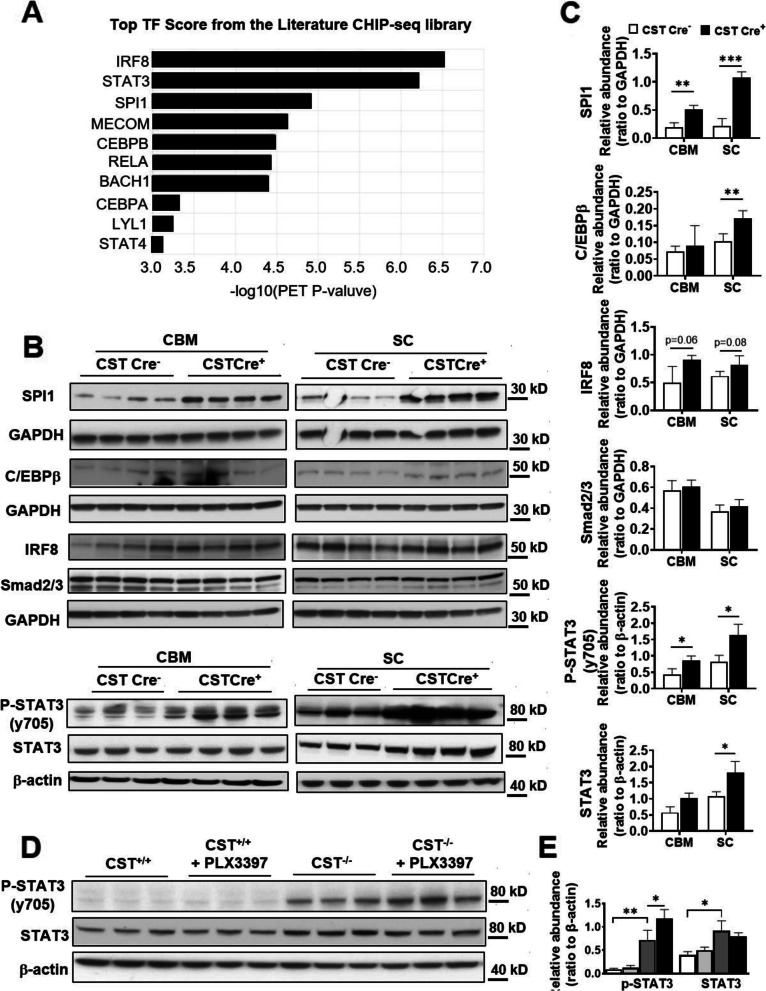
Results: We found that mild central nervous system (CNS) sulfatide losses within myelinating cells are sufficient to activate disease-associated microglia and astrocytes, and to increase the expression of AD risk genes (e.g., Apoe, Trem2, Cd33, and Mmp12), as well as previously established causal regulators of the immune/microglia network in late-onset AD (e.g., Tyrobp, Dock, and Fcerg1), leading to chronic AD-like neuroinflammation and mild cognitive impairment. Notably, neuroinflammation and mild cognitive impairment showed gender differences, being more pronounced in females than males. Subsequent mechanistic studies demonstrated that although CNS sulfatide losses led to ApoE upregulation, genetically-induced myelin sulfatide deficiency led to neuroinflammation independently of ApoE. These results, together with our previous studies (sulfatide deficiency in the context of AD is mediated by ApoE and accelerated by Aβ accumulation) placed both Aβ and ApoE upstream of sulfatide deficiency-induced neuroinflammation, and suggested a positive feedback loop where sulfatide losses may be amplified by increased ApoE expression. We also demonstrated that CNS sulfatide deficiency-induced astrogliosis and ApoE upregulation are not secondary to microgliosis, and that astrogliosis and microgliosis seem to be driven by activation of STAT3 and PU.1/Spi1 transcription factors, respectively.
Conclusion: Our results strongly suggest that sulfatide deficiency is an important contributor and driver of neuroinflammation and mild cognitive impairment in AD pathology.
Keywords: Alzheimer’s disease; Astrogliosis; Cerebroside sulfotransferase (CST); Cognitive impairment; Lipidomics; Microgliosis; Neuroinflammation; RNA profiling; Sulfatide.
© 2021. The Author(s).
Conflict of interest statement
Authors declare that they have no competing interests.
Figures


Similar articles
-
Novel molecular insights into the critical role of sulfatide in myelin maintenance/function.J Neurochem. 2016 Oct;139(1):40-54. doi: 10.1111/jnc.13738. Epub 2016 Aug 15. J Neurochem. 2016. PMID: 27417284 Free PMC article.
-
Glial Sulfatides and Neuronal Complex Gangliosides Are Functionally Interdependent in Maintaining Myelinating Axon Integrity.J Neurosci. 2019 Jan 2;39(1):63-77. doi: 10.1523/JNEUROSCI.2095-18.2018. Epub 2018 Nov 16. J Neurosci. 2019. PMID: 30446529 Free PMC article.
-
Apolipoprotein E mediates sulfatide depletion in animal models of Alzheimer's disease.Neurobiol Aging. 2010 Jul;31(7):1188-96. doi: 10.1016/j.neurobiolaging.2008.07.020. Epub 2008 Aug 31. Neurobiol Aging. 2010. PMID: 18762354 Free PMC article.
-
The pathogenic implication of abnormal interaction between apolipoprotein E isoforms, amyloid-beta peptides, and sulfatides in Alzheimer's disease.Mol Neurobiol. 2010 Jun;41(2-3):97-106. doi: 10.1007/s12035-009-8092-x. Epub 2010 Jan 7. Mol Neurobiol. 2010. PMID: 20052565 Free PMC article. Review.
-
Potential mechanisms contributing to sulfatide depletion at the earliest clinically recognizable stage of Alzheimer's disease: a tale of shotgun lipidomics.J Neurochem. 2007 Nov;103 Suppl 1(Suppl 1):171-9. doi: 10.1111/j.1471-4159.2007.04708.x. J Neurochem. 2007. PMID: 17986152 Free PMC article. Review.
Cited by
-
Deterioration of neuroimmune homeostasis in Alzheimer's Disease patients who survive a COVID-19 infection.J Neuroinflammation. 2024 Aug 17;21(1):202. doi: 10.1186/s12974-024-03196-3. J Neuroinflammation. 2024. PMID: 39154174 Free PMC article.
-
Adult-onset depletion of sulfatide leads to axonal degeneration with relative myelin sparing.Glia. 2023 Sep;71(9):2285-2303. doi: 10.1002/glia.24423. Epub 2023 Jun 7. Glia. 2023. PMID: 37283058 Free PMC article.
-
Gut microbiota metabolites: potential therapeutic targets for Alzheimer's disease?Front Pharmacol. 2024 Sep 17;15:1459655. doi: 10.3389/fphar.2024.1459655. eCollection 2024. Front Pharmacol. 2024. PMID: 39355779 Free PMC article. Review.
-
Dietary calcium intake at breakfast is associated with a lower risk of cognitive impairment than at dinner in Chinese adults: the CHNS cohort study.Asia Pac J Clin Nutr. 2024 Sep;33(3):405-412. doi: 10.6133/apjcn.202409_33(3).0011. Asia Pac J Clin Nutr. 2024. PMID: 38965728 Free PMC article.
-
Myelinodegeneration vs. Neurodegeneration in MS Progressive Forms.Int J Mol Sci. 2023 Jan 13;24(2):1596. doi: 10.3390/ijms24021596. Int J Mol Sci. 2023. PMID: 36675111 Free PMC article.
References
-
- Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14(4):388–405. doi: 10.1016/S1474-4422(15)70016-5. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
