

Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome
- PMID: 33891876
- PMCID: PMC8152331
- DOI: 10.1016/j.cell.2021.03.048
Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome
Abstract
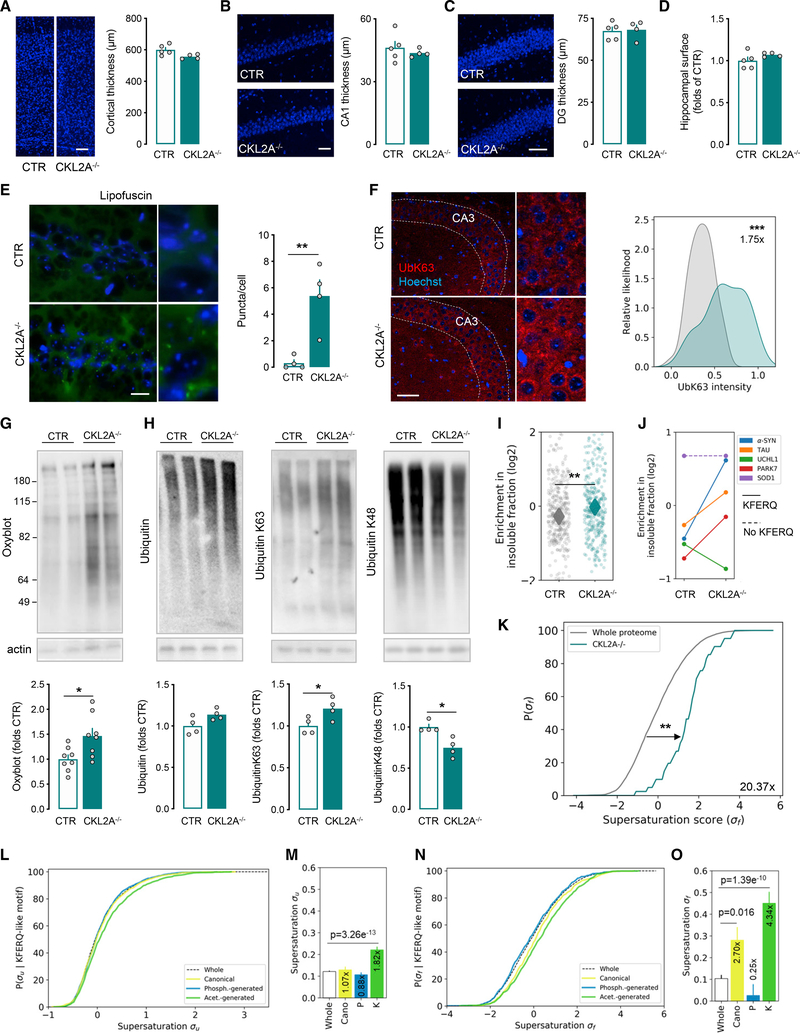
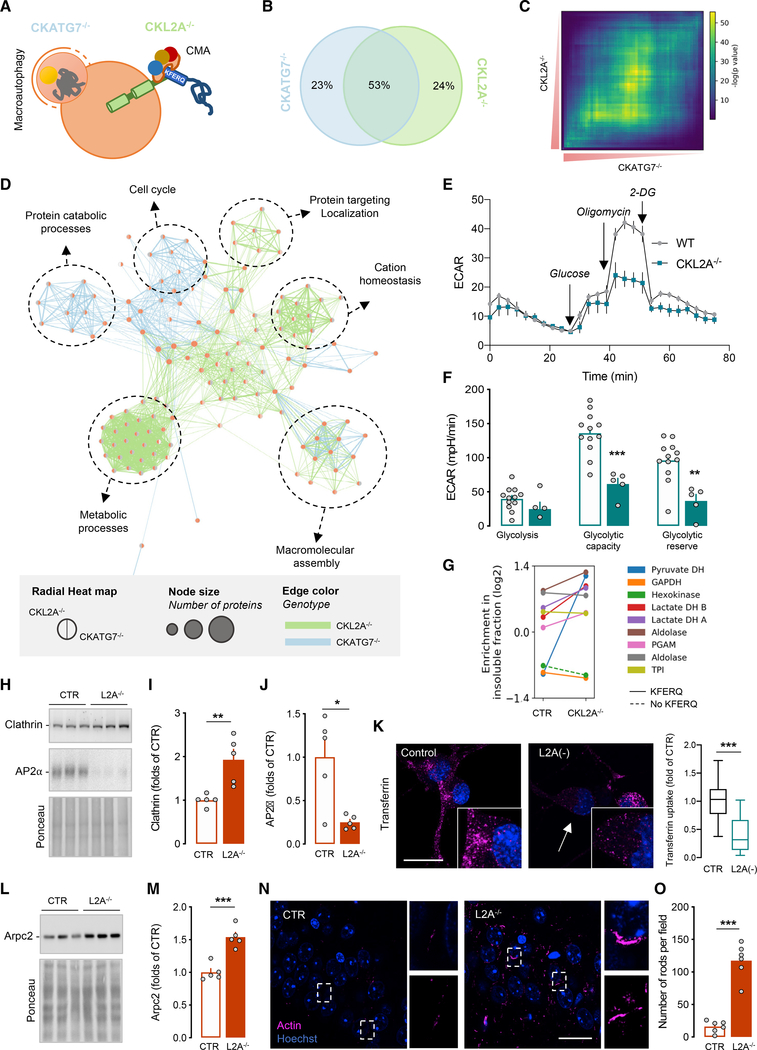
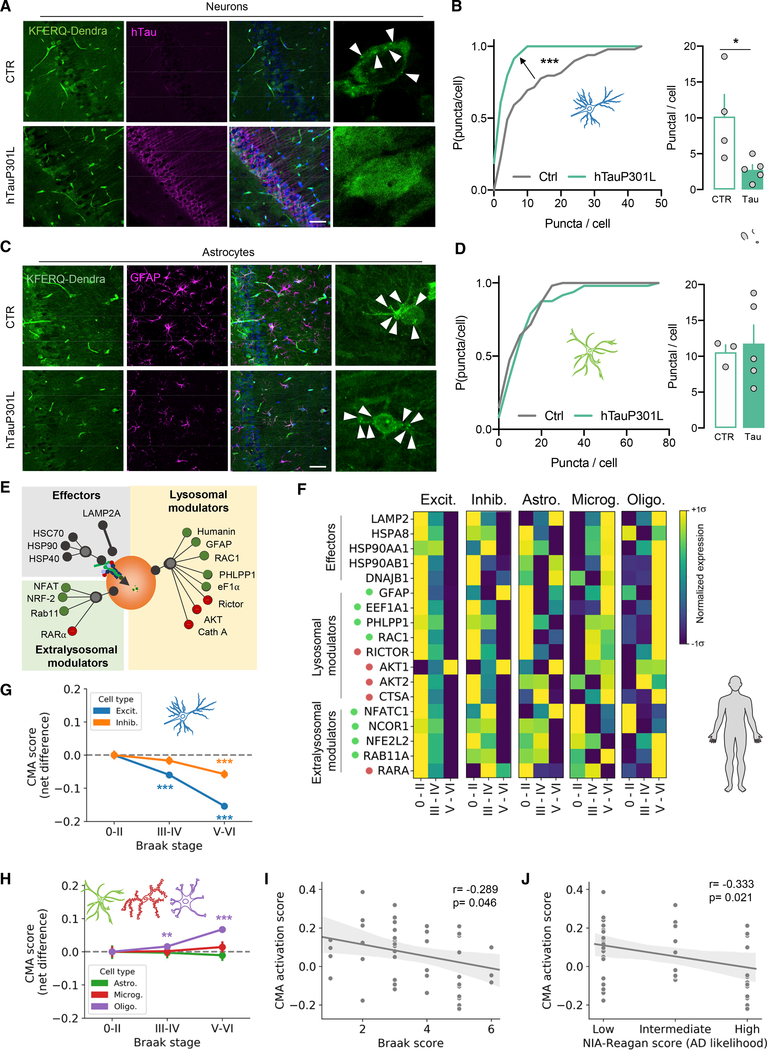
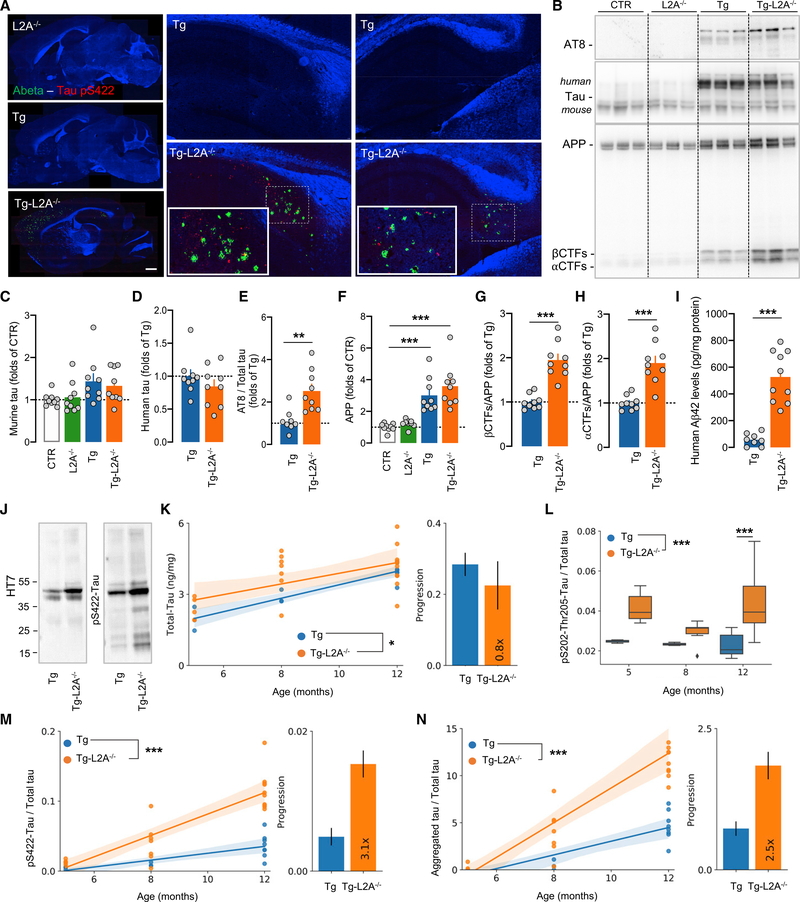
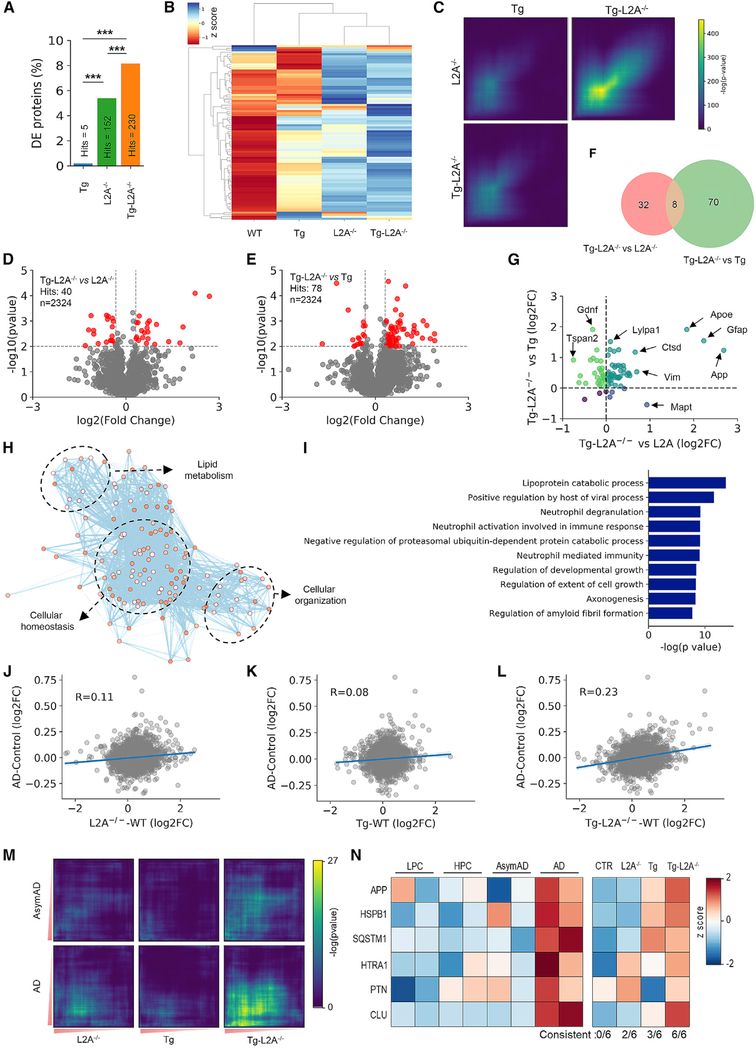
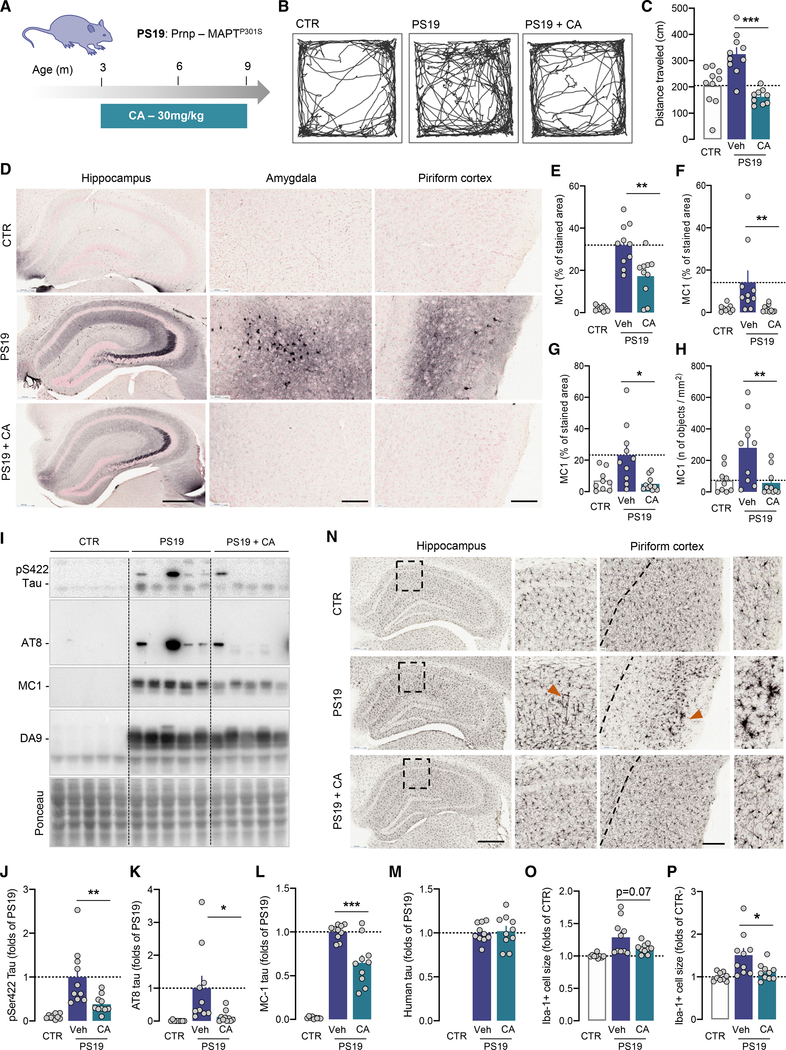
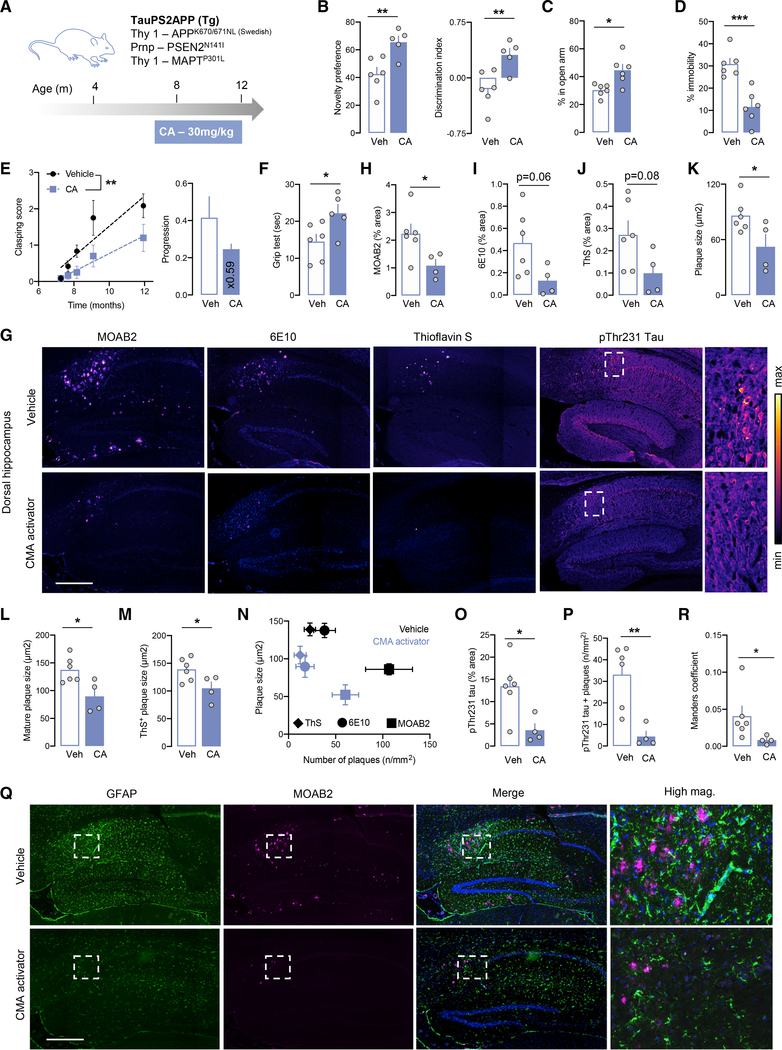
Components of the proteostasis network malfunction in aging, and reduced protein quality control in neurons has been proposed to promote neurodegeneration. Here, we investigate the role of chaperone-mediated autophagy (CMA), a selective autophagy shown to degrade neurodegeneration-related proteins, in neuronal proteostasis. Using mouse models with systemic and neuronal-specific CMA blockage, we demonstrate that loss of neuronal CMA leads to altered neuronal function, selective changes in the neuronal metastable proteome, and proteotoxicity, all reminiscent of brain aging. Imposing CMA loss on a mouse model of Alzheimer's disease (AD) has synergistic negative effects on the proteome at risk of aggregation, thus increasing neuronal disease vulnerability and accelerating disease progression. Conversely, chemical enhancement of CMA ameliorates pathology in two different AD experimental mouse models. We conclude that functional CMA is essential for neuronal proteostasis through the maintenance of a subset of the proteome with a higher risk of misfolding than the general proteome.
Keywords: Alzheimer’s disease; aging; chaperones; chemical activators of autophagy; lysosomes; neurodegeneration; protein aggregation; proteotoxicity; supersaturated proteome; tau.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.M.C. and E.G. are cofounders and scientific advisors for Life Biosciences. A.M.C. consults for Generian Pharmaceuticals and Cognition Therapeutics. N.J.K. consults for Maze Therapeutics and Interline Therapeutics, received research support from Vir Biotechnology and F. Hoffmann-La Roche, and is a shareholder of Tenaya Therapeutics. The remaining authors declare no competing interests. CA compound is under US patent US9512092 (E.G., A.M.C., and Q.X.).
Figures
Similar articles
-
Chaperone-mediated autophagy: a gatekeeper of neuronal proteostasis.Autophagy. 2021 Aug;17(8):2040-2042. doi: 10.1080/15548627.2021.1935007. Epub 2021 Jun 10. Autophagy. 2021. PMID: 34110247 Free PMC article. Review.
-
Neurons die with heightened but functional macro- and chaperone mediated autophagy upon increased amyloid-ß induced toxicity with region-specific protection in prolonged intermittent fasting.Exp Cell Res. 2021 Nov 15;408(2):112840. doi: 10.1016/j.yexcr.2021.112840. Epub 2021 Oct 9. Exp Cell Res. 2021. PMID: 34624324
-
Calorie restriction and calorie-restriction mimetics activate chaperone-mediated autophagy.Proc Natl Acad Sci U S A. 2024 Jun 25;121(26):e2317945121. doi: 10.1073/pnas.2317945121. Epub 2024 Jun 18. Proc Natl Acad Sci U S A. 2024. PMID: 38889154 Free PMC article.
-
Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging.Aging Cell. 2015 Apr;14(2):249-64. doi: 10.1111/acel.12310. Epub 2015 Jan 23. Aging Cell. 2015. PMID: 25620427 Free PMC article.
-
Chaperone-mediated autophagy in neurodegenerative diseases: mechanisms and therapy.Mol Cell Biochem. 2023 Oct;478(10):2173-2190. doi: 10.1007/s11010-022-04640-9. Epub 2023 Jan 25. Mol Cell Biochem. 2023. PMID: 36695937 Review.
Cited by
-
Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases.Nat Rev Mol Cell Biol. 2024 Nov;25(11):926-946. doi: 10.1038/s41580-024-00757-5. Epub 2024 Aug 6. Nat Rev Mol Cell Biol. 2024. PMID: 39107446 Review.
-
Hypothesis-based investigation of known AD risk variants reveals the genetic underpinnings of neuropathological lesions observed in Alzheimer's-type dementia.Acta Neuropathol. 2024 Oct 18;148(1):55. doi: 10.1007/s00401-024-02815-w. Acta Neuropathol. 2024. PMID: 39424714 Free PMC article.
-
Chaperone-Mediated Autophagy in Neurodegenerative Diseases and Acute Neurological Insults in the Central Nervous System.Cells. 2022 Apr 2;11(7):1205. doi: 10.3390/cells11071205. Cells. 2022. PMID: 35406769 Free PMC article. Review.
-
Palmitic and Stearic Acids Inhibit Chaperone-Mediated Autophagy (CMA) in POMC-like Neurons In Vitro.Cells. 2022 Mar 8;11(6):920. doi: 10.3390/cells11060920. Cells. 2022. PMID: 35326371 Free PMC article.
-
Chaperone-Mediated Autophagy in Neurodegenerative Diseases: Molecular Mechanisms and Pharmacological Opportunities.Cells. 2022 Jul 20;11(14):2250. doi: 10.3390/cells11142250. Cells. 2022. PMID: 35883693 Free PMC article. Review.
References
-
- Aniento F, Roche E, Cuervo AM, and Knecht E (1993). Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 268, 10463–10470. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
