

Revealing the Activity of Trimeric G-proteins in Live Cells with a Versatile Biosensor Design
- PMID: 32634377
- PMCID: PMC7415655
- DOI: 10.1016/j.cell.2020.06.020
Revealing the Activity of Trimeric G-proteins in Live Cells with a Versatile Biosensor Design
Abstract
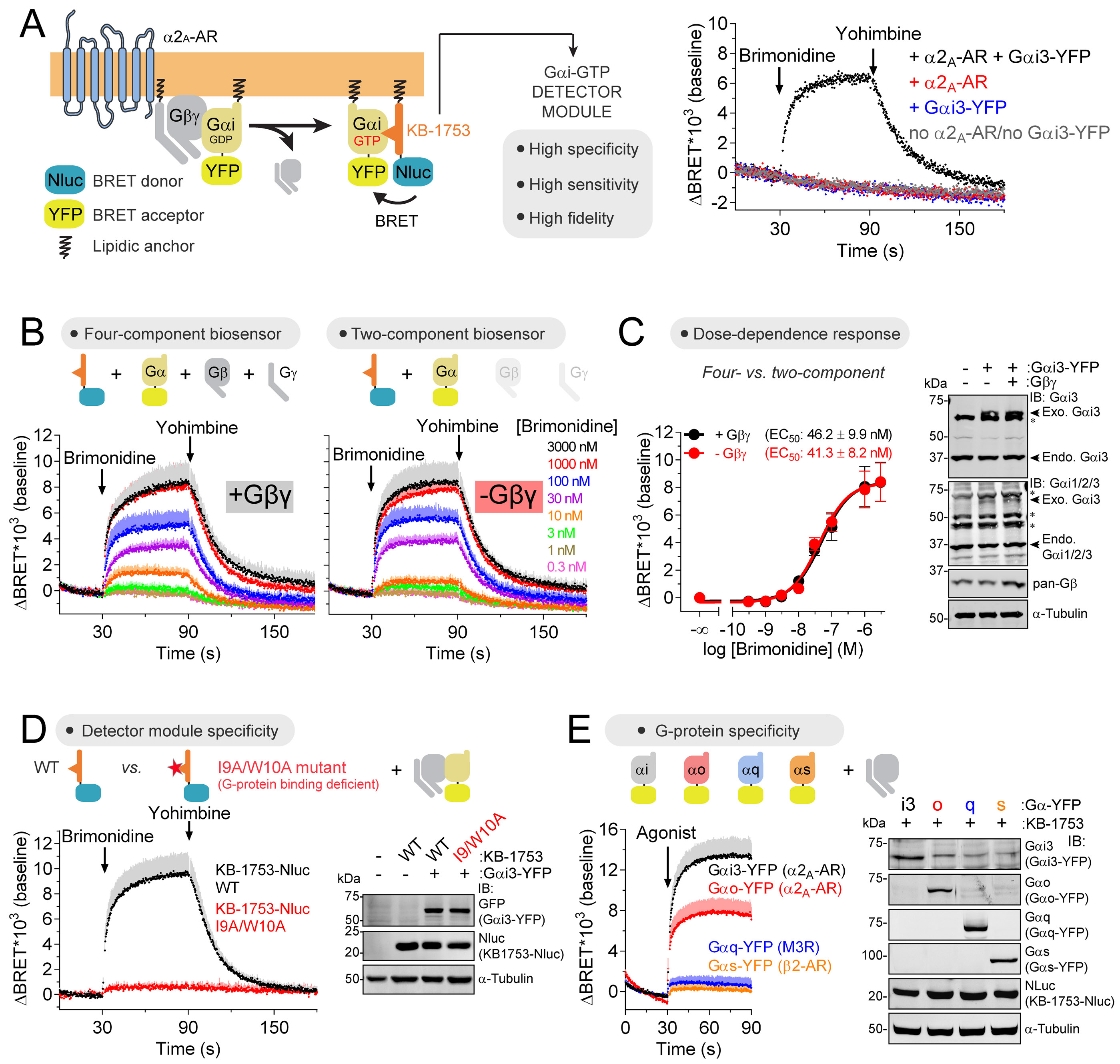
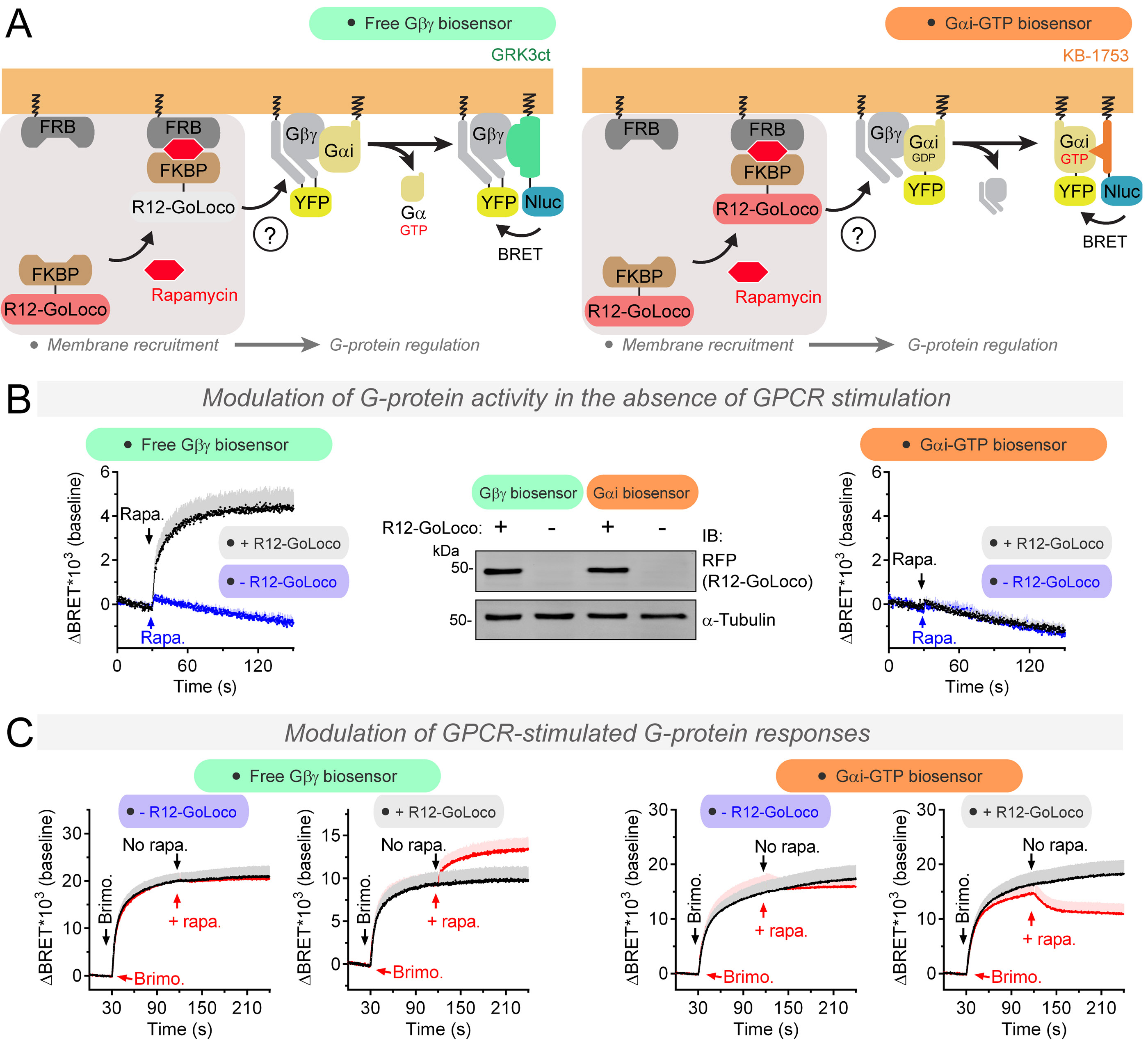
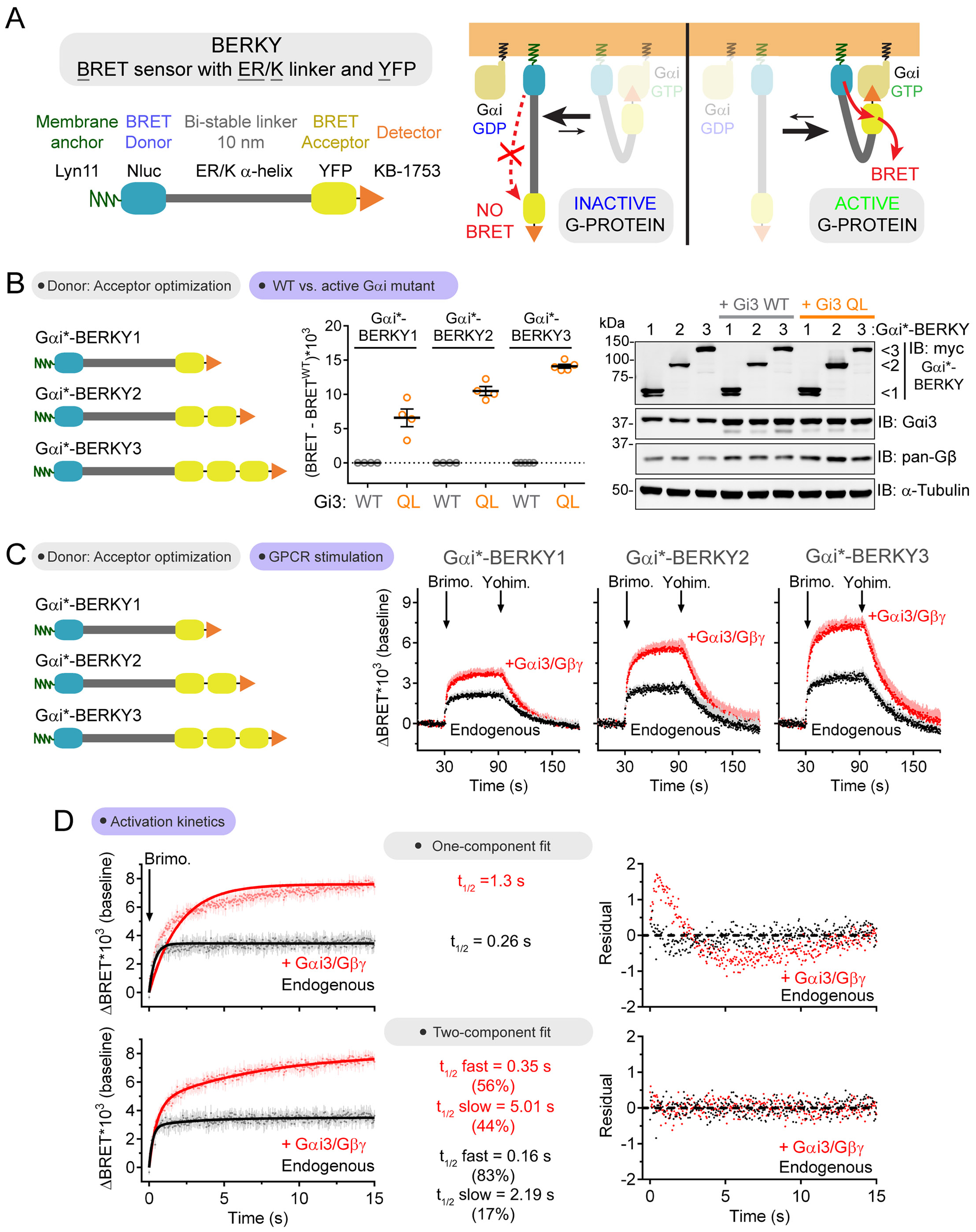
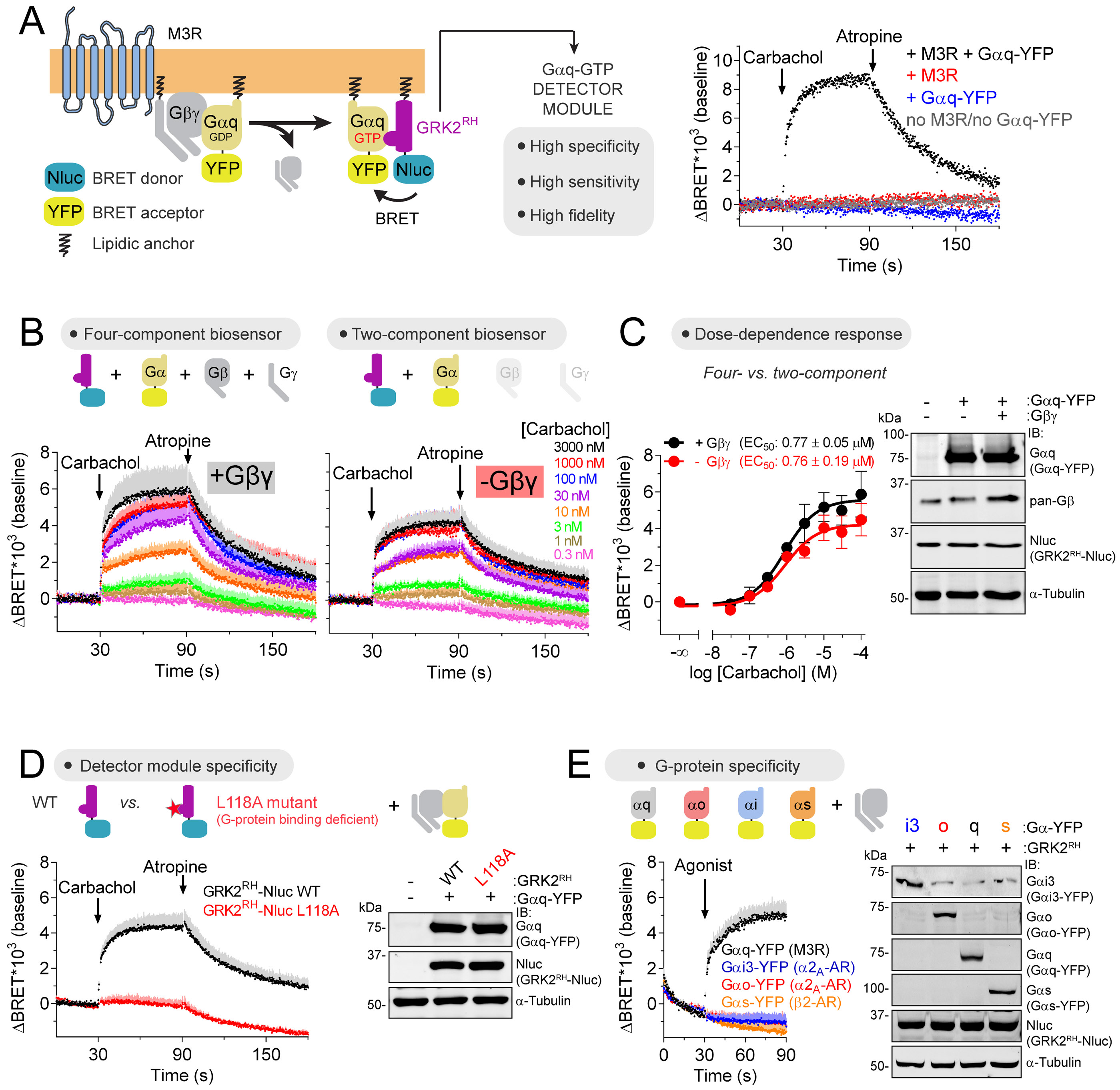
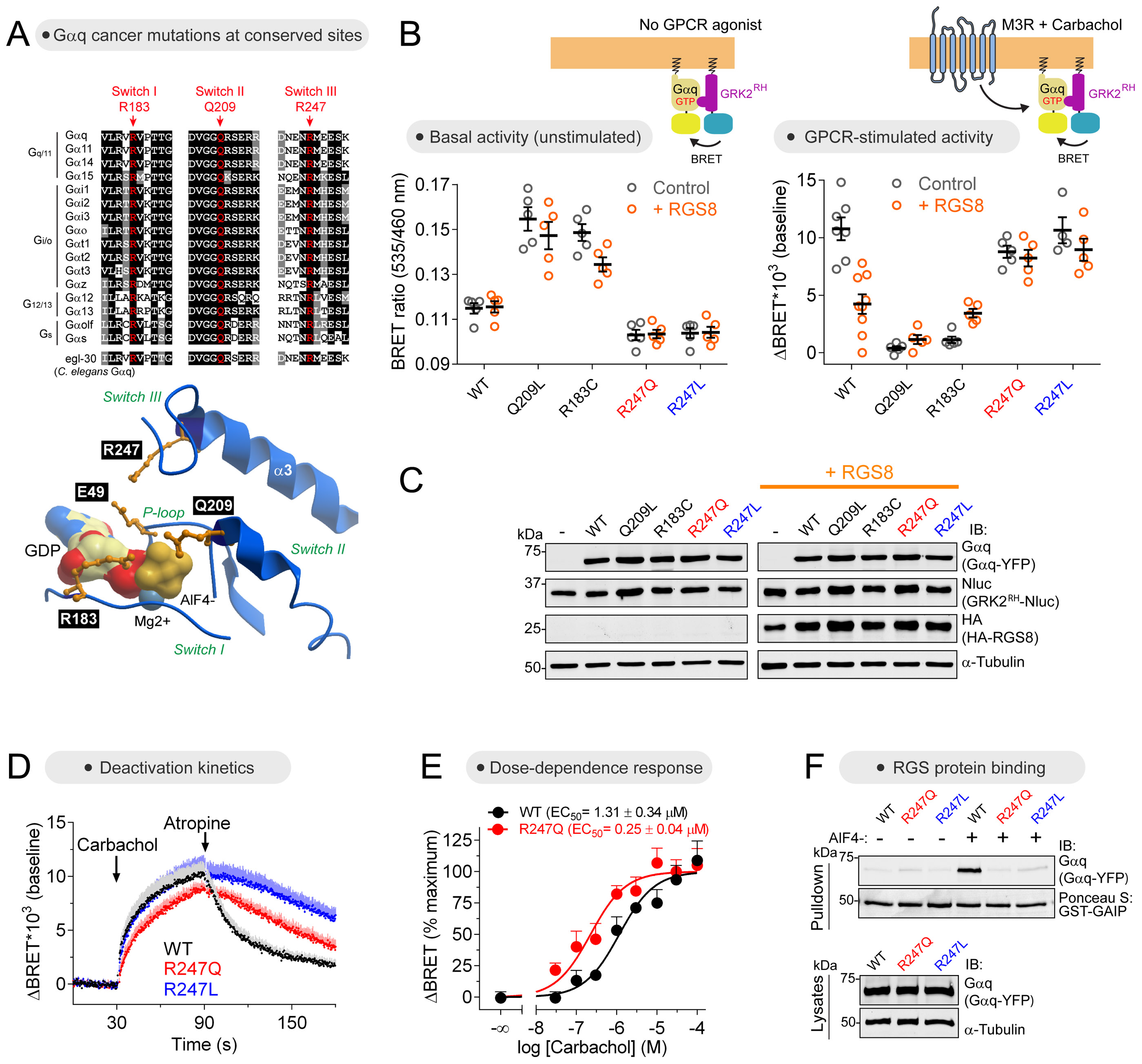
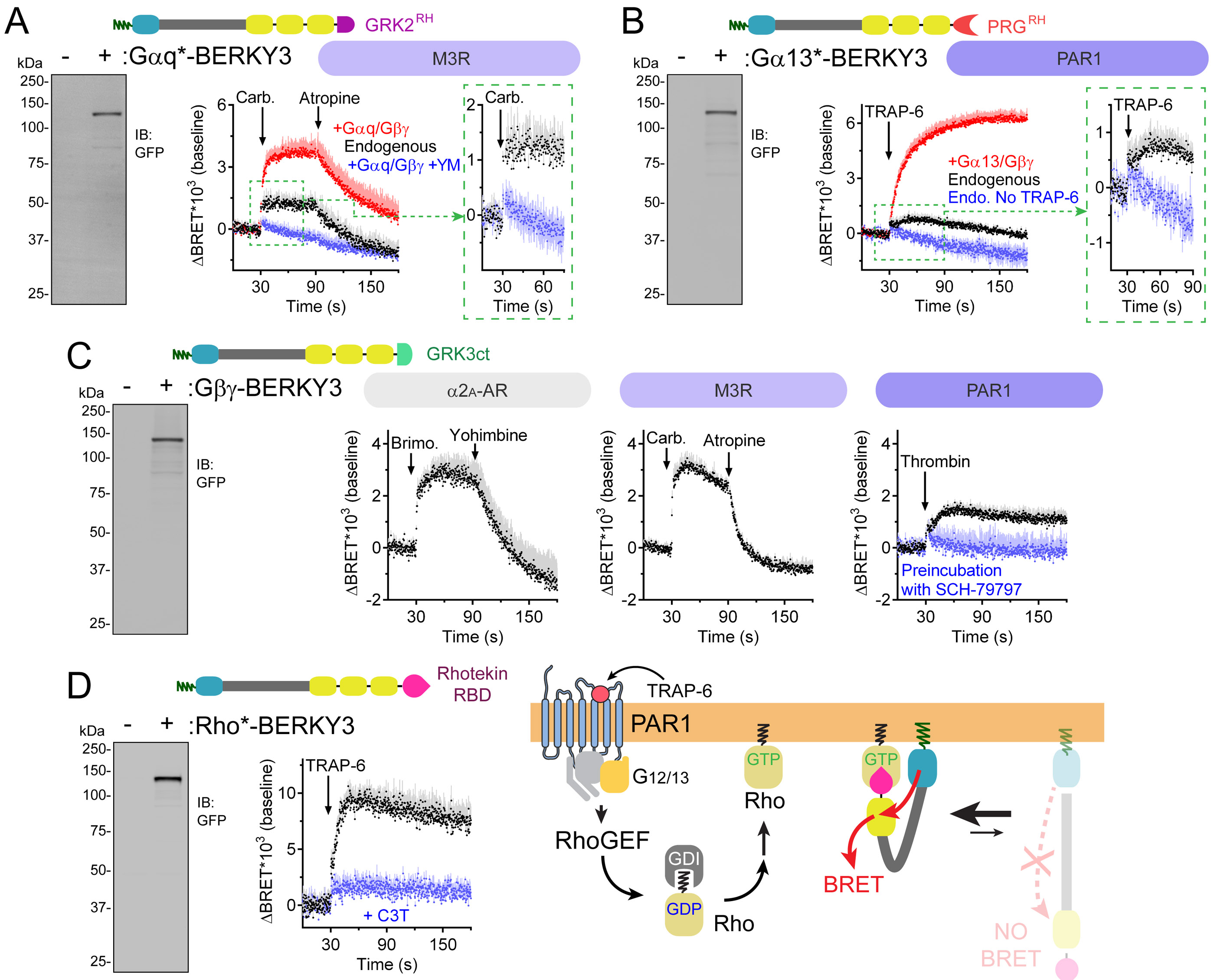
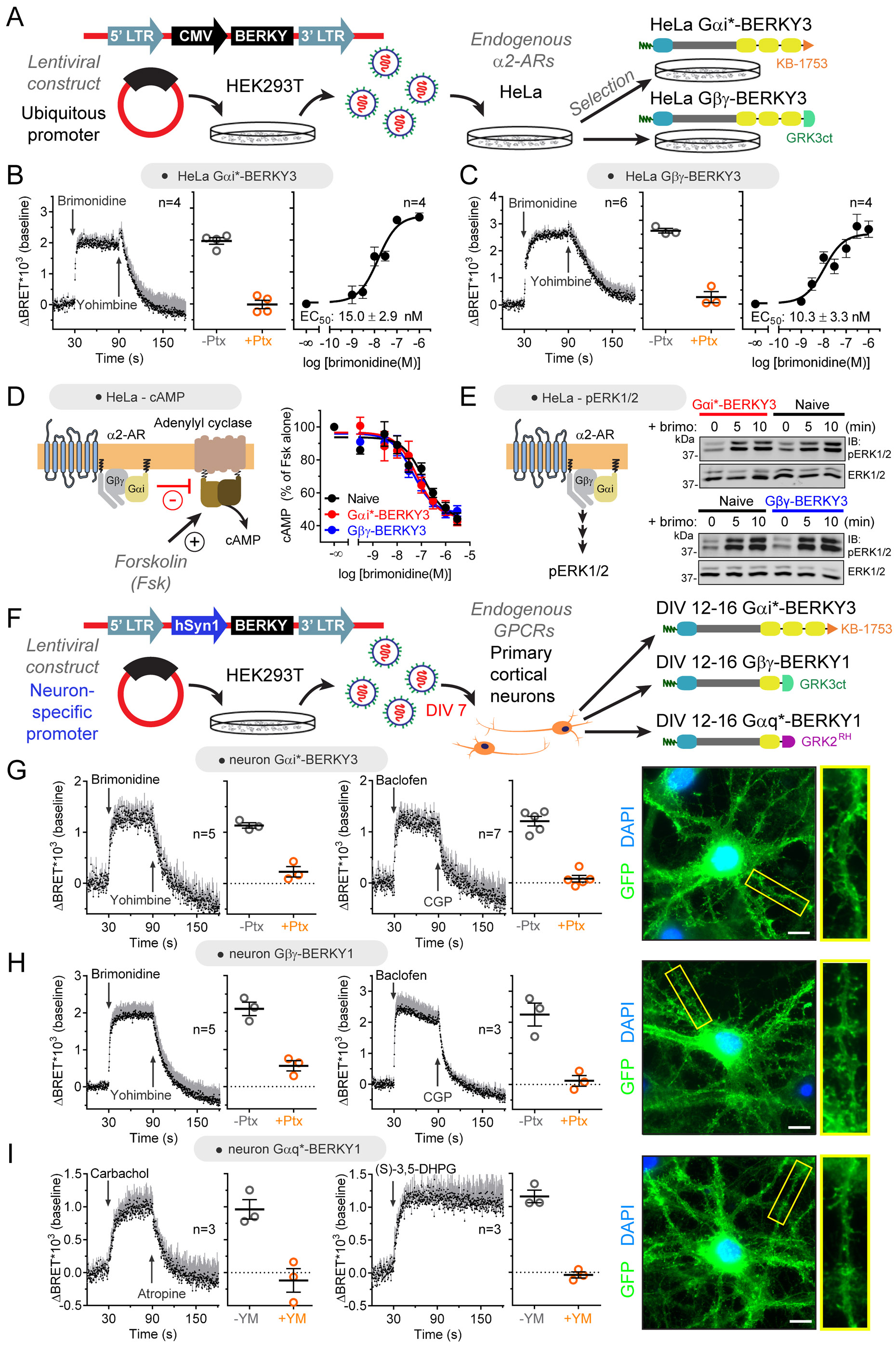
Heterotrimeric G-proteins (Gαβγ) are the main transducers of signals from GPCRs, mediating the action of countless natural stimuli and therapeutic agents. However, there are currently no robust approaches to directly measure the activity of endogenous G-proteins in cells. Here, we describe a suite of optical biosensors that detect endogenous active G-proteins with sub-second resolution in live cells. Using a modular design principle, we developed genetically encoded, unimolecular biosensors for endogenous Gα-GTP and free Gβγ: the two active species of heterotrimeric G-proteins. This design was leveraged to generate biosensors with specificity for different heterotrimeric G-proteins or for other G-proteins, such as Rho GTPases. Versatility was further validated by implementing the biosensors in multiple contexts, from characterizing cancer-associated G-protein mutants to neurotransmitter signaling in primary neurons. Overall, the versatile biosensor design introduced here enables studying the activity of endogenous G-proteins in live cells with high fidelity, temporal resolution, and convenience.
Keywords: BRET; G protein; GPCR; GTPase; biosensor; cancer; neurobiology; neurotransmitter; oncogene.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests M.G.-M. is listed as an inventor in a provisional patent filed by Boston University related to the content of this manuscript.
Figures
Similar articles
-
Detecting GPCR Signals With Optical Biosensors of Gα-GTP in Cell Lines and Primary Cell Cultures.Curr Protoc. 2023 Jun;3(6):e796. doi: 10.1002/cpz1.796. Curr Protoc. 2023. PMID: 37310083 Free PMC article.
-
Rapid kinetic BRET measurements to monitor G protein activation by GPCR and non-GPCR proteins.Methods Cell Biol. 2017;142:145-157. doi: 10.1016/bs.mcb.2017.08.001. Epub 2017 Sep 12. Methods Cell Biol. 2017. PMID: 28964333 Free PMC article.
-
Complementary biosensors reveal different G-protein signaling modes triggered by GPCRs and non-receptor activators.Elife. 2021 Mar 31;10:e65620. doi: 10.7554/eLife.65620. Elife. 2021. PMID: 33787494 Free PMC article.
-
The GAPs, GEFs, and GDIs of heterotrimeric G-protein alpha subunits.Int J Biol Sci. 2005;1(2):51-66. doi: 10.7150/ijbs.1.51. Epub 2005 Apr 1. Int J Biol Sci. 2005. PMID: 15951850 Free PMC article. Review.
-
Emerging non-canonical functions for heterotrimeric G proteins in cellular signaling.J Recept Signal Transduct Res. 2013 Jun;33(3):177-83. doi: 10.3109/10799893.2013.795972. J Recept Signal Transduct Res. 2013. PMID: 23721574 Review.
Cited by
-
Investigating G-protein coupled receptor signalling with light-emitting biosensors.Front Physiol. 2024 Jan 8;14:1310197. doi: 10.3389/fphys.2023.1310197. eCollection 2023. Front Physiol. 2024. PMID: 38260094 Free PMC article. Review.
-
Direct interrogation of context-dependent GPCR activity with a universal biosensor platform.bioRxiv [Preprint]. 2024 Jan 2:2024.01.02.573921. doi: 10.1101/2024.01.02.573921. bioRxiv. 2024. Update in: Cell. 2024 Mar 14;187(6):1527-1546.e25. doi: 10.1016/j.cell.2024.01.028 PMID: 38260348 Free PMC article. Updated. Preprint.
-
Genetically encoded fluorescent biosensors for GPCR research.Front Cell Dev Biol. 2022 Sep 29;10:1007893. doi: 10.3389/fcell.2022.1007893. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36247000 Free PMC article. Review.
-
Neomorphic Gαo mutations gain interaction with Ric8 proteins in GNAO1 encephalopathies.J Clin Invest. 2024 Jun 14;134(15):e172057. doi: 10.1172/JCI172057. J Clin Invest. 2024. PMID: 38874642 Free PMC article.
-
Detecting and measuring of GPCR signaling - comparison of human induced pluripotent stem cells and immortal cell lines.Front Endocrinol (Lausanne). 2023 May 24;14:1179600. doi: 10.3389/fendo.2023.1179600. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 37293485 Free PMC article. Review.
References
-
- Berman DM, Wilkie TM, and Gilman AG (1996). GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell 86, 445–452. - PubMed
-
- Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, and Kozasa T (1999). Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. The Journal of biological chemistry 274, 34483–34492. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
