

Uncovering tissue-specific binding features from differential deep learning
- PMID: 31974574
- PMCID: PMC7049686
- DOI: 10.1093/nar/gkaa009
Uncovering tissue-specific binding features from differential deep learning
Abstract
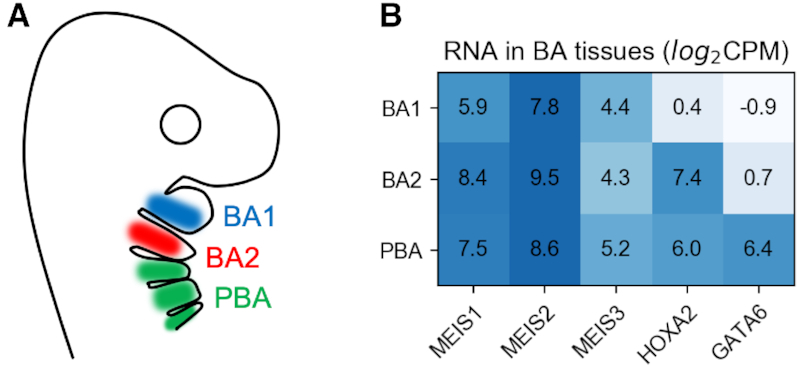
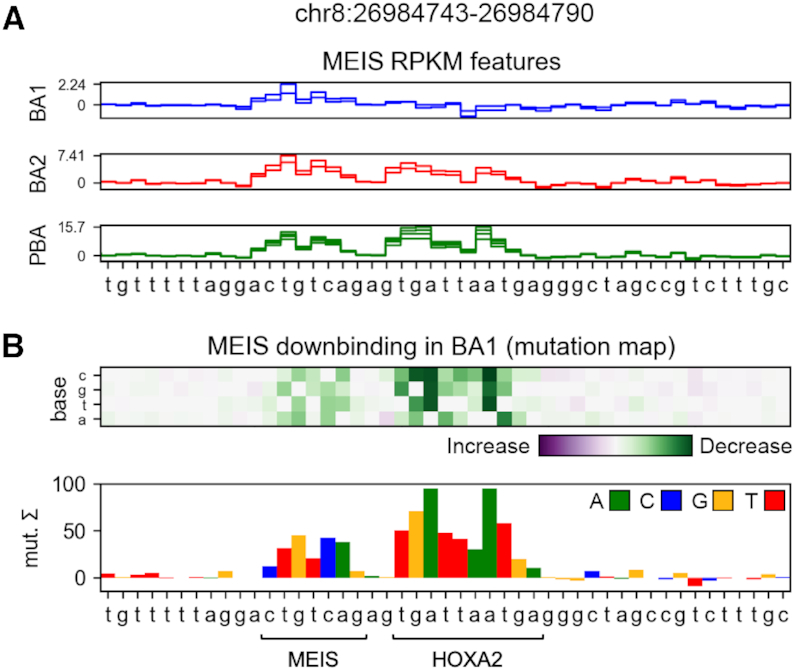
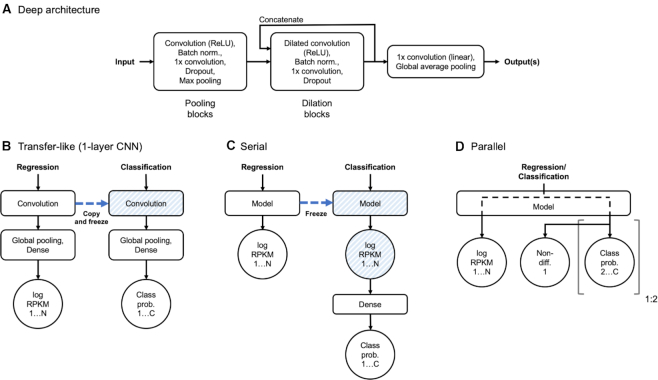
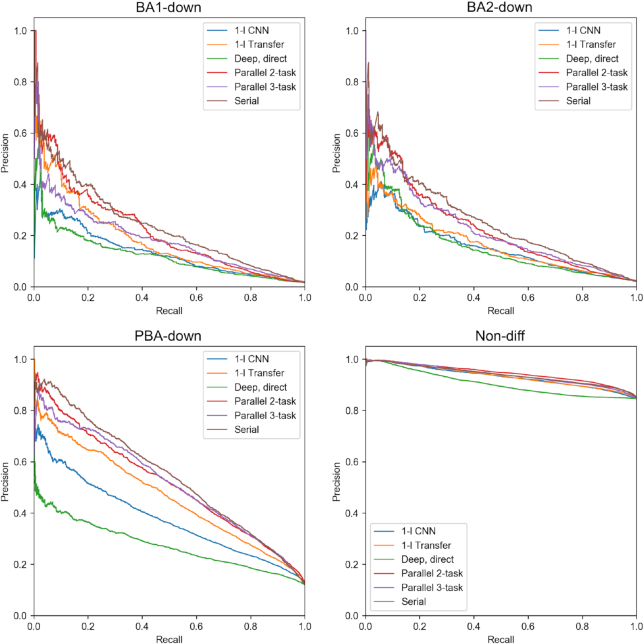
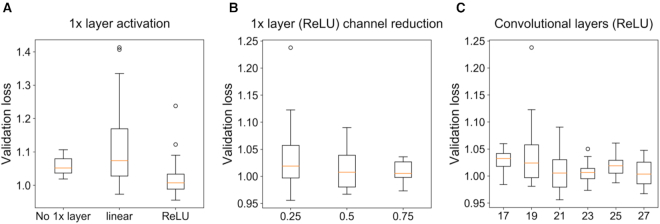
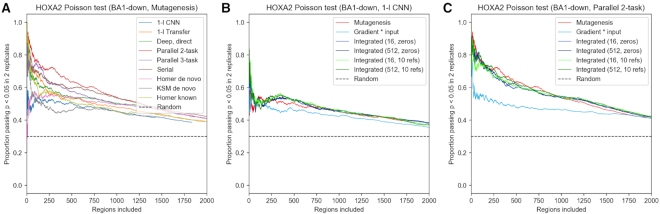
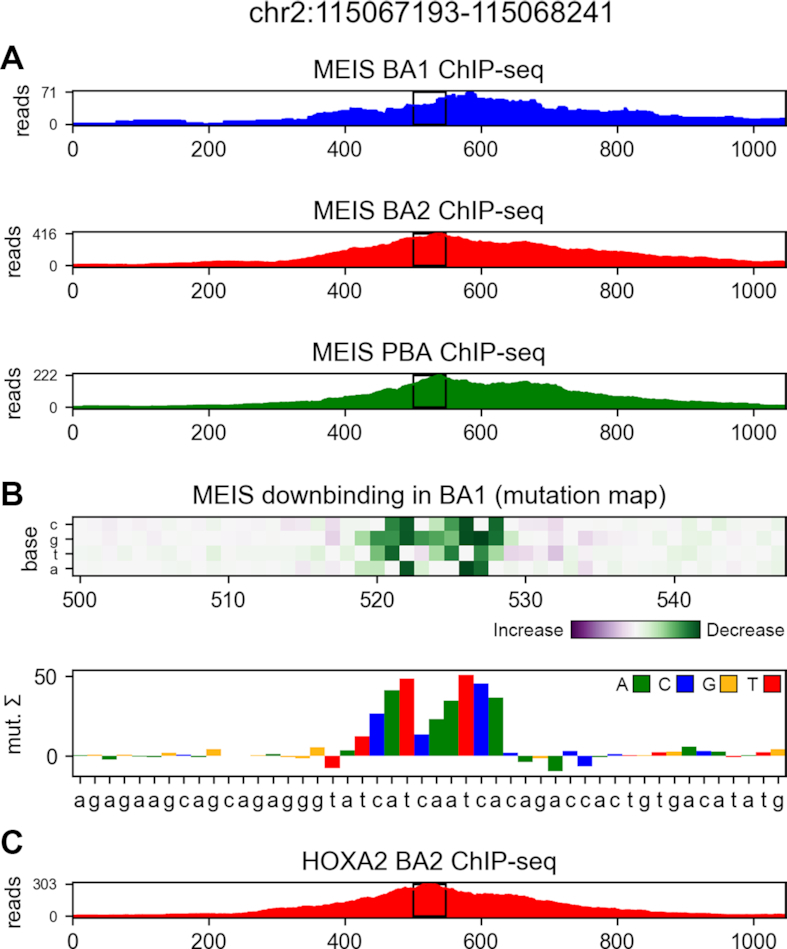
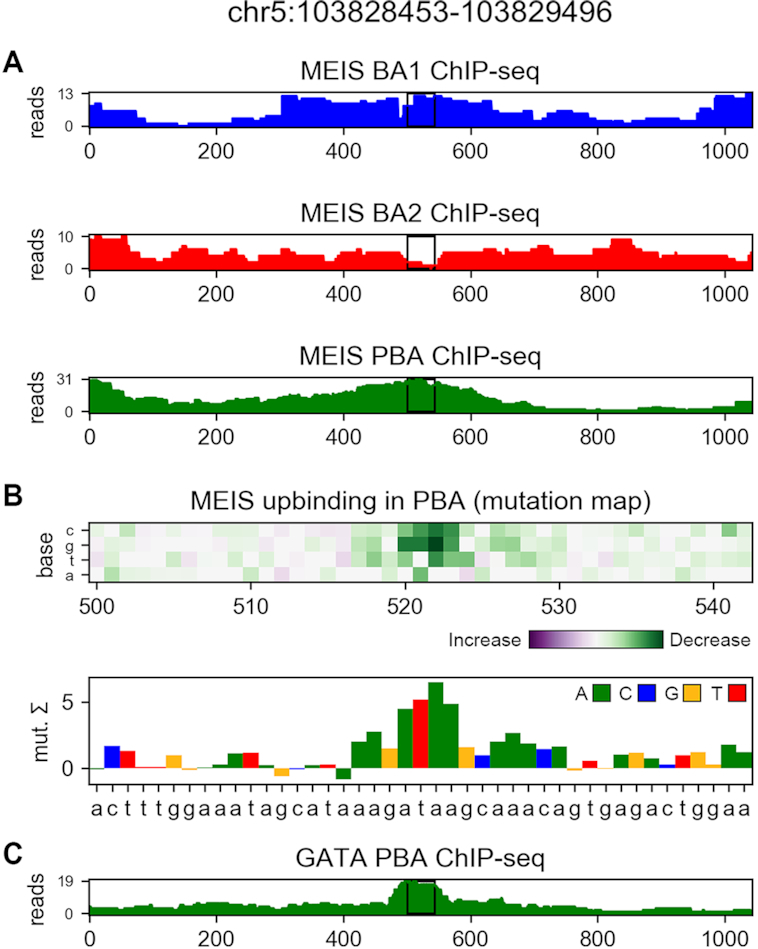
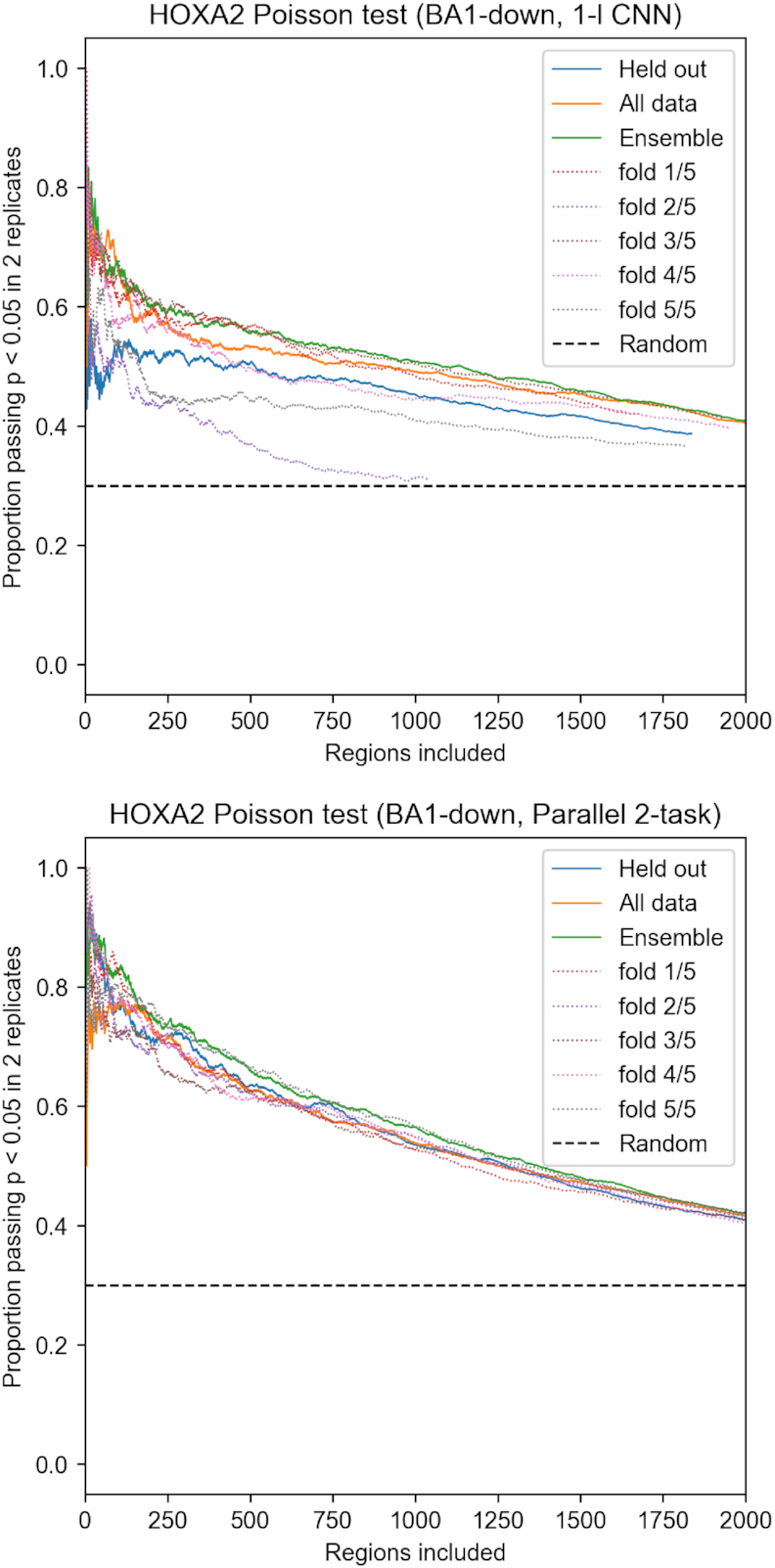
Transcription factors (TFs) can bind DNA in a cooperative manner, enabling a mutual increase in occupancy. Through this type of interaction, alternative binding sites can be preferentially bound in different tissues to regulate tissue-specific expression programmes. Recently, deep learning models have become state-of-the-art in various pattern analysis tasks, including applications in the field of genomics. We therefore investigate the application of convolutional neural network (CNN) models to the discovery of sequence features determining cooperative and differential TF binding across tissues. We analyse ChIP-seq data from MEIS, TFs which are broadly expressed across mouse branchial arches, and HOXA2, which is expressed in the second and more posterior branchial arches. By developing models predictive of MEIS differential binding in all three tissues, we are able to accurately predict HOXA2 co-binding sites. We evaluate transfer-like and multitask approaches to regularizing the high-dimensional classification task with a larger regression dataset, allowing for the creation of deeper and more accurate models. We test the performance of perturbation and gradient-based attribution methods in identifying the HOXA2 sites from differential MEIS data. Our results show that deep regularized models significantly outperform shallow CNNs as well as k-mer methods in the discovery of tissue-specific sites bound in vivo.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Hoxa2 selectively enhances Meis binding to change a branchial arch ground state.Dev Cell. 2015 Feb 9;32(3):265-77. doi: 10.1016/j.devcel.2014.12.024. Epub 2015 Jan 29. Dev Cell. 2015. PMID: 25640223 Free PMC article.
-
Differential distribution of the Ca (2+) regulator Pcp4 in the branchial arches is regulated by Hoxa2.PLoS One. 2013 May 9;8(5):e63160. doi: 10.1371/journal.pone.0063160. Print 2013. PLoS One. 2013. PMID: 23671666 Free PMC article.
-
Transient activation of meox1 is an early component of the gene regulatory network downstream of hoxa2.Mol Cell Biol. 2011 Mar;31(6):1301-8. doi: 10.1128/MCB.00705-10. Epub 2011 Jan 18. Mol Cell Biol. 2011. PMID: 21245383 Free PMC article.
-
The regulatory landscape of the Dlx gene system in branchial arches: Shared characteristics among Dlx bigene clusters and evolution.Dev Growth Differ. 2020 Jun;62(5):355-362. doi: 10.1111/dgd.12671. Epub 2020 May 30. Dev Growth Differ. 2020. PMID: 32403166 Review.
-
Utility of next-generation RNA-sequencing in identifying chimeric transcription involving human endogenous retroviruses.APMIS. 2016 Jan-Feb;124(1-2):127-39. doi: 10.1111/apm.12477. APMIS. 2016. PMID: 26818267 Review.
Cited by
-
HOX paralogs selectively convert binding of ubiquitous transcription factors into tissue-specific patterns of enhancer activation.PLoS Genet. 2020 Dec 14;16(12):e1009162. doi: 10.1371/journal.pgen.1009162. eCollection 2020 Dec. PLoS Genet. 2020. PMID: 33315856 Free PMC article.
-
Host-pathogen protein-nucleic acid interactions: A comprehensive review.Comput Struct Biotechnol J. 2022 Aug 4;20:4415-4436. doi: 10.1016/j.csbj.2022.08.001. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36051878 Free PMC article. Review.
-
JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles.Nucleic Acids Res. 2024 Jan 5;52(D1):D174-D182. doi: 10.1093/nar/gkad1059. Nucleic Acids Res. 2024. PMID: 37962376 Free PMC article.
-
Identifying transcription factors with cell-type specific DNA binding signatures.BMC Genomics. 2024 Oct 14;25(1):957. doi: 10.1186/s12864-024-10859-1. BMC Genomics. 2024. PMID: 39402535 Free PMC article.
-
Transcription factor-binding k-mer analysis clarifies the cell type dependency of binding specificities and cis-regulatory SNPs in humans.BMC Genomics. 2023 Oct 7;24(1):597. doi: 10.1186/s12864-023-09692-9. BMC Genomics. 2023. PMID: 37805453 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
