

TGFβ-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis
- PMID: 31243287
- PMCID: PMC6595054
- DOI: 10.1038/s41467-019-10677-0
TGFβ-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis
Abstract
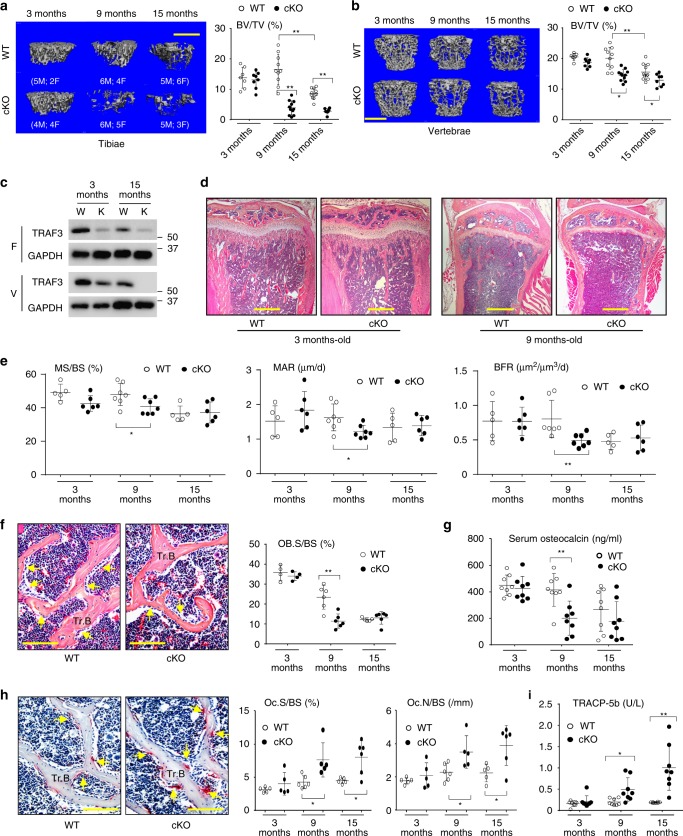
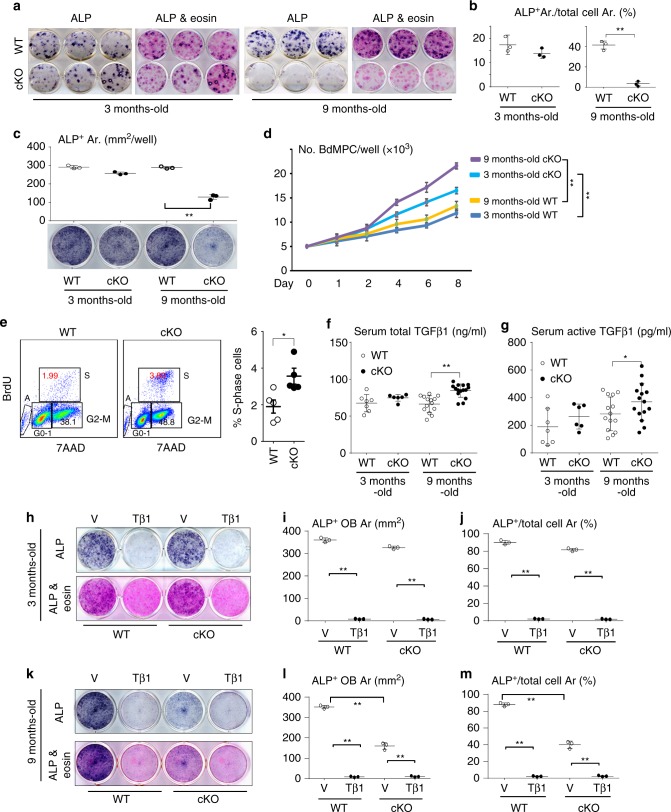
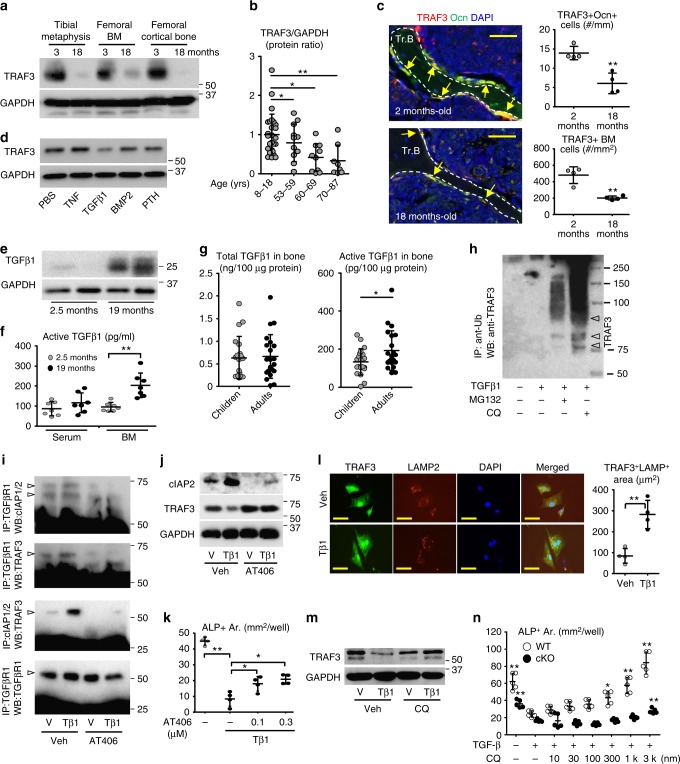
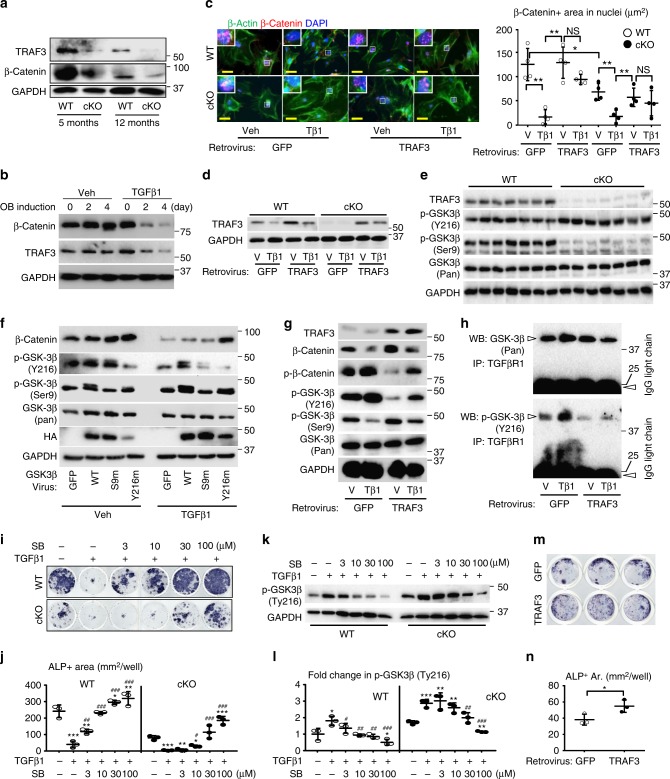
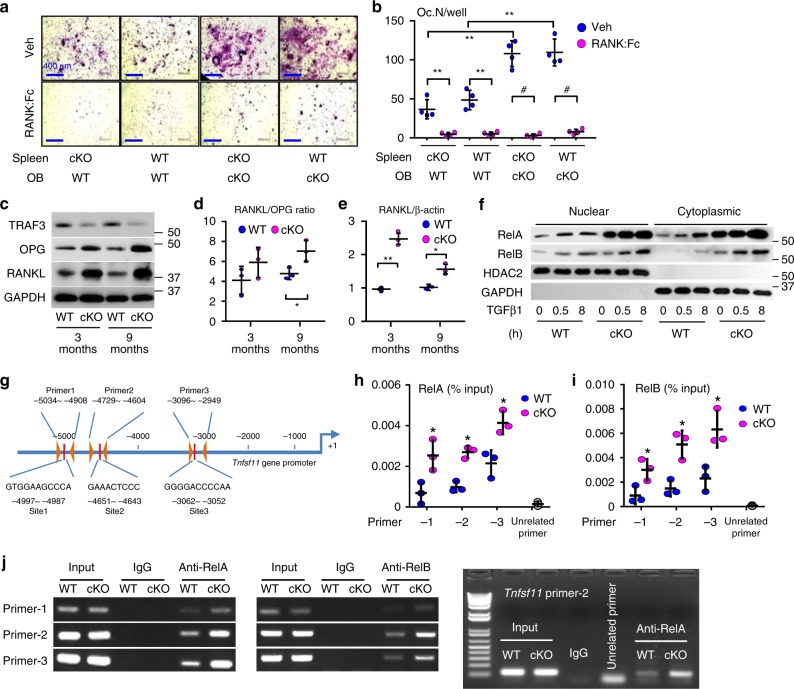
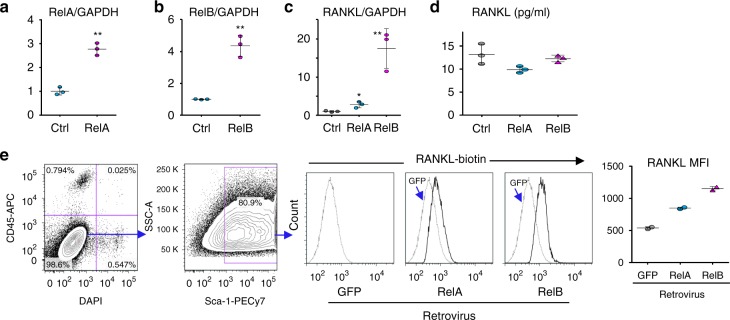
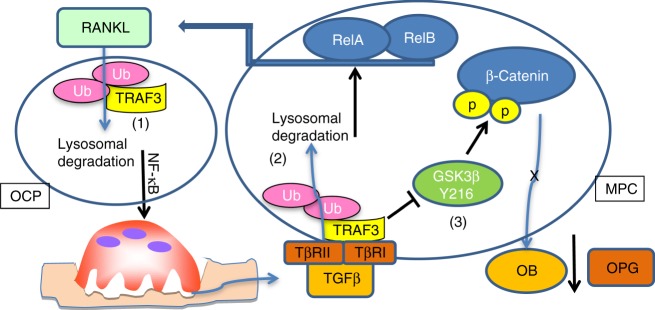
Inflammaging induces osteoporosis by promoting bone destruction and inhibiting bone formation. TRAF3 limits bone destruction by inhibiting RANKL-induced NF-κB signaling in osteoclast precursors. However, the role of TRAF3 in mesenchymal progenitor cells (MPCs) is unknown. Mice with TRAF3 deleted in MPCs develop early onset osteoporosis due to reduced bone formation and enhanced bone destruction. In young mice TRAF3 prevents β-catenin degradation in MPCs and maintains osteoblast formation. However, TRAF3 protein levels decrease in murine and human bone samples during aging when TGFβ1 is released from resorbing bone. TGFβ1 induces degradation of TRAF3 in murine MPCs and inhibits osteoblast formation through GSK-3β-mediated degradation of β-catenin. Thus, TRAF3 positively regulates MPC differentiation into osteoblasts. TRAF3 deletion in MPCs activated NF-κB RelA and RelB to promote RANKL expression and enhance bone destruction. We conclude that pharmacologic stabilization of TRAF3 during aging could treat/prevent age-related osteoporosis by inhibiting bone destruction and promoting bone formation.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Nuclear Factor-Kappa B Regulation of Osteoclastogenesis and Osteoblastogenesis.Endocrinol Metab (Seoul). 2023 Oct;38(5):504-521. doi: 10.3803/EnM.2023.501. Epub 2023 Sep 26. Endocrinol Metab (Seoul). 2023. PMID: 37749800 Free PMC article.
-
Bone Remodeling and the Role of TRAF3 in Osteoclastic Bone Resorption.Front Immunol. 2018 Sep 28;9:2263. doi: 10.3389/fimmu.2018.02263. eCollection 2018. Front Immunol. 2018. PMID: 30323820 Free PMC article. Review.
-
TGFβ1+CCR5+ neutrophil subset increases in bone marrow and causes age-related osteoporosis in male mice.Nat Commun. 2023 Jan 11;14(1):159. doi: 10.1038/s41467-023-35801-z. Nat Commun. 2023. PMID: 36631487 Free PMC article.
-
Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation.J Clin Invest. 2014 Jan;124(1):297-310. doi: 10.1172/JCI66947. Epub 2013 Dec 9. J Clin Invest. 2014. PMID: 24316970 Free PMC article.
-
Regulation of TNF-Induced Osteoclast Differentiation.Cells. 2021 Dec 31;11(1):132. doi: 10.3390/cells11010132. Cells. 2021. PMID: 35011694 Free PMC article. Review.
Cited by
-
Integrated Strategy of Network Pharmacological Prediction and Experimental Validation Elucidate Possible Mechanism of Bu-Yang Herbs in Treating Postmenopausal Osteoporosis via ESR1.Front Pharmacol. 2021 May 11;12:654714. doi: 10.3389/fphar.2021.654714. eCollection 2021. Front Pharmacol. 2021. PMID: 34045964 Free PMC article.
-
Vascular Pericyte-Derived Exosomes Inhibit Bone Resorption via Traf3.Int J Nanomedicine. 2023 Nov 28;18:7065-7077. doi: 10.2147/IJN.S438229. eCollection 2023. Int J Nanomedicine. 2023. PMID: 38046234 Free PMC article.
-
Extracellular Vesicles in Bone Homeostasis: Emerging Mediators of Osteoimmune Interactions and Promising Therapeutic Targets.Int J Biol Sci. 2022 Jun 21;18(10):4088-4100. doi: 10.7150/ijbs.69816. eCollection 2022. Int J Biol Sci. 2022. PMID: 35844790 Free PMC article. Review.
-
TNF Receptor-Associated Factor 6 Mediates TNFα-Induced Skeletal Muscle Atrophy in Mice During Aging.J Bone Miner Res. 2020 Aug;35(8):1535-1548. doi: 10.1002/jbmr.4021. Epub 2020 Apr 27. J Bone Miner Res. 2020. PMID: 32267572 Free PMC article.
-
Osteoclasts and osteoarthritis: Novel intervention targets and therapeutic potentials during aging.Aging Cell. 2024 Apr;23(4):e14092. doi: 10.1111/acel.14092. Epub 2024 Jan 29. Aging Cell. 2024. PMID: 38287696 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
