

Structure-guided fragment-based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping
- PMID: 31030650
- PMCID: PMC6501894
- DOI: 10.1098/rsta.2018.0422
Structure-guided fragment-based drug discovery at the synchrotron: screening binding sites and correlations with hotspot mapping
Abstract
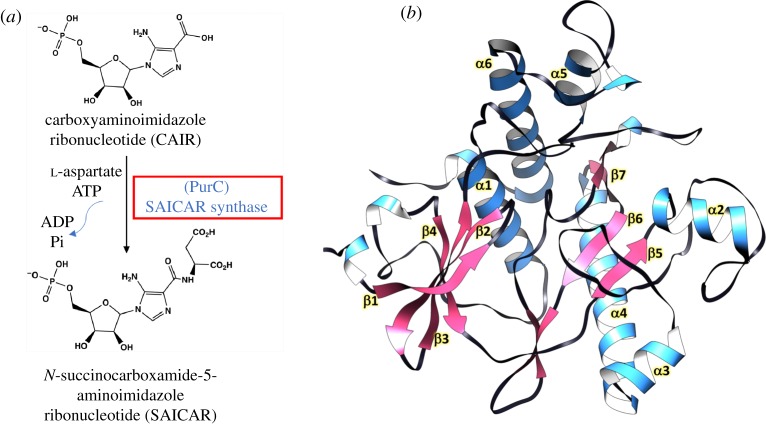
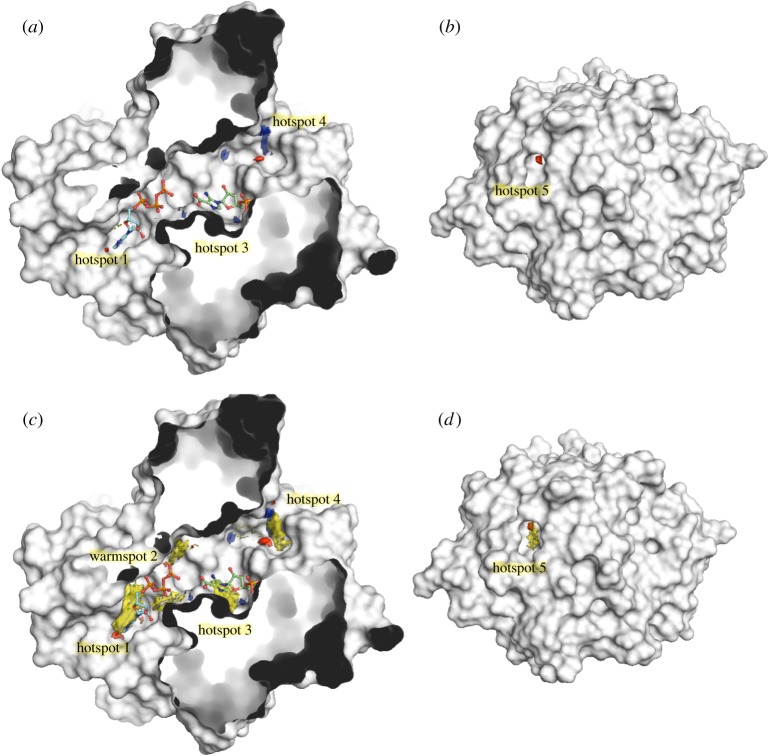
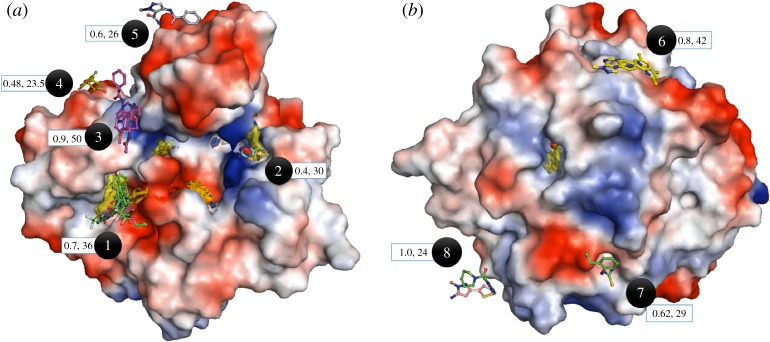
Structure-guided drug discovery emerged in the 1970s and 1980s, stimulated by the three-dimensional structures of protein targets that became available, mainly through X-ray crystal structure analysis, assisted by the development of synchrotron radiation sources. Structures of known drugs or inhibitors were used to guide the development of leads. The growth of high-throughput screening during the late 1980s and the early 1990s in the pharmaceutical industry of chemical libraries of hundreds of thousands of compounds of molecular weight of approximately 500 Da was impressive but still explored only a tiny fraction of the chemical space of the predicted 1040 drug-like compounds. The use of fragments with molecular weights less than 300 Da in drug discovery not only decreased the chemical space needing exploration but also increased promiscuity in binding targets. Here we discuss advances in X-ray fragment screening and the challenge of identifying sites where fragments not only bind but can be chemically elaborated while retaining their positions and binding modes. We first describe the analysis of fragment binding using conventional X-ray difference Fourier techniques, with Mycobacterium abscessus SAICAR synthetase (PurC) as an example. We observe that all fragments occupy positions predicted by computational hotspot mapping. We compare this with fragment screening at Diamond Synchrotron Light Source XChem facility using PanDDA software, which identifies many more fragment hits, only some of which bind to the predicted hotspots. Many low occupancy sites identified may not support elaboration to give adequate ligand affinity, although they will likely be useful in drug discovery as 'warm spots' for guiding elaboration of fragments bound at hotspots. We discuss implications of these observations for fragment screening at the synchrotron sources. This article is part of the theme issue 'Fifty years of synchrotron science: achievements and opportunities'.
Keywords: Mycobacterium abscessus; SAICAR synthetase (PurC); fragment-based drug discovery; structure-guided; synchrotron.
Conflict of interest statement
We declare we have no competing interests.
Figures

Similar articles
-
Development of Inhibitors of SAICAR Synthetase (PurC) from Mycobacterium abscessus Using a Fragment-Based Approach.ACS Infect Dis. 2022 Feb 11;8(2):296-309. doi: 10.1021/acsinfecdis.1c00432. Epub 2022 Jan 17. ACS Infect Dis. 2022. PMID: 35037462 Free PMC article.
-
Integrated biophysical approach to fragment screening and validation for fragment-based lead discovery.Proc Natl Acad Sci U S A. 2013 Aug 6;110(32):12984-9. doi: 10.1073/pnas.1304045110. Epub 2013 Jul 19. Proc Natl Acad Sci U S A. 2013. PMID: 23872845 Free PMC article.
-
Fragment-based approaches in drug discovery and chemical biology.Biochemistry. 2012 Jun 26;51(25):4990-5003. doi: 10.1021/bi3005126. Epub 2012 Jun 14. Biochemistry. 2012. PMID: 22697260 Review.
-
Fast fragment and compound screening pipeline at the Swiss Light Source.Methods Enzymol. 2023;690:235-284. doi: 10.1016/bs.mie.2023.08.005. Epub 2023 Sep 12. Methods Enzymol. 2023. PMID: 37858531 Review.
-
Fragments: where are we now?Biochem Soc Trans. 2020 Feb 28;48(1):271-280. doi: 10.1042/BST20190694. Biochem Soc Trans. 2020. PMID: 31985743 Review.
Cited by
-
Fragment-Based Drug Discovery against Mycobacteria: The Success and Challenges.Int J Mol Sci. 2022 Sep 14;23(18):10669. doi: 10.3390/ijms231810669. Int J Mol Sci. 2022. PMID: 36142582 Free PMC article. Review.
-
Fragment-based discovery of a new class of inhibitors targeting mycobacterial tRNA modification.Nucleic Acids Res. 2020 Aug 20;48(14):8099-8112. doi: 10.1093/nar/gkaa539. Nucleic Acids Res. 2020. PMID: 32602532 Free PMC article.
-
Management of Mycobacterium avium complex and Mycobacterium abscessus pulmonary disease: therapeutic advances and emerging treatments.Eur Respir Rev. 2022 Feb 9;31(163):210212. doi: 10.1183/16000617.0212-2021. Print 2022 Mar 31. Eur Respir Rev. 2022. PMID: 35140106 Free PMC article. Review.
-
Ligand-centered assessment of SARS-CoV-2 drug target models in the Protein Data Bank.FEBS J. 2020 Sep;287(17):3703-3718. doi: 10.1111/febs.15366. Epub 2020 Jun 24. FEBS J. 2020. PMID: 32418327 Free PMC article.
-
Perspective: Structure determination of protein-ligand complexes at room temperature using X-ray diffraction approaches.Front Mol Biosci. 2023 Jan 23;10:1113762. doi: 10.3389/fmolb.2023.1113762. eCollection 2023. Front Mol Biosci. 2023. PMID: 36756363 Free PMC article.
References
-
- Thomas SE, Mendes V, Kim SY, Malhotra S, Ochoa-Montano B, Blaszczyk M, Blundell TL. 2017. Structural biology and the design of new therapeutics: from HIV and cancer to mycobacterial infections: a paper dedicated to John Kendrew. J. Mol. Biol. 429, 2677–2693. (10.1016/j.jmb.2017.06.014) - DOI - PubMed
-
- Subramanian E, Swan ID, Liu M, Davies DR, Jenkins JA, Tickle IJ, Blundell TL. 1977. Homology among acid proteases: comparison of crystal structures at 3A resolution of acid proteases from Rhizopus chinensis and Endothia parasitica. Proc. Natl Acad. Sci. USA 74, 556–559. (10.1073/pnas.74.2.556) - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
