

Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval
- PMID: 29381698
- PMCID: PMC5806898
- DOI: 10.1371/journal.pbio.2004411
Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval
Abstract
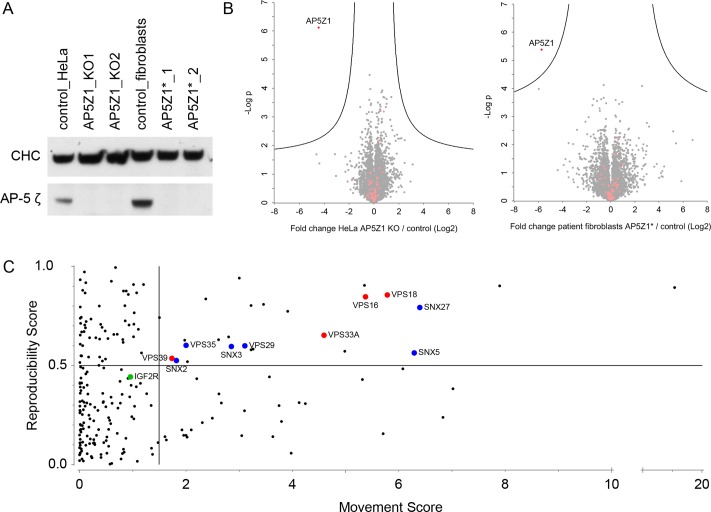
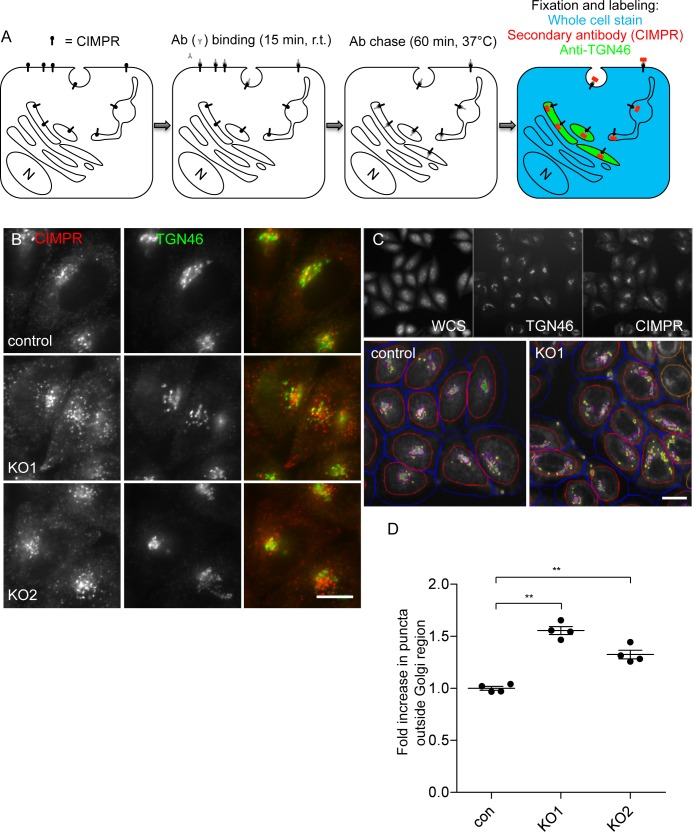
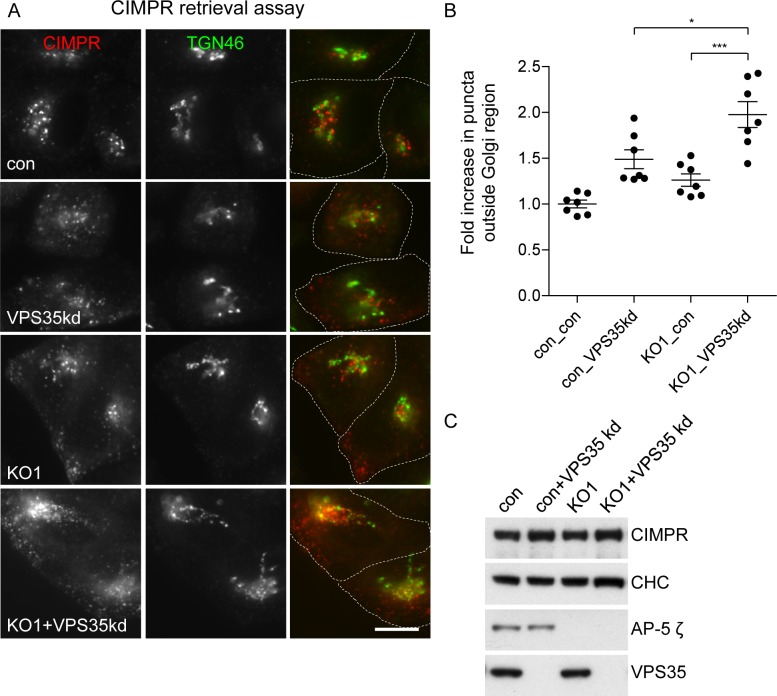
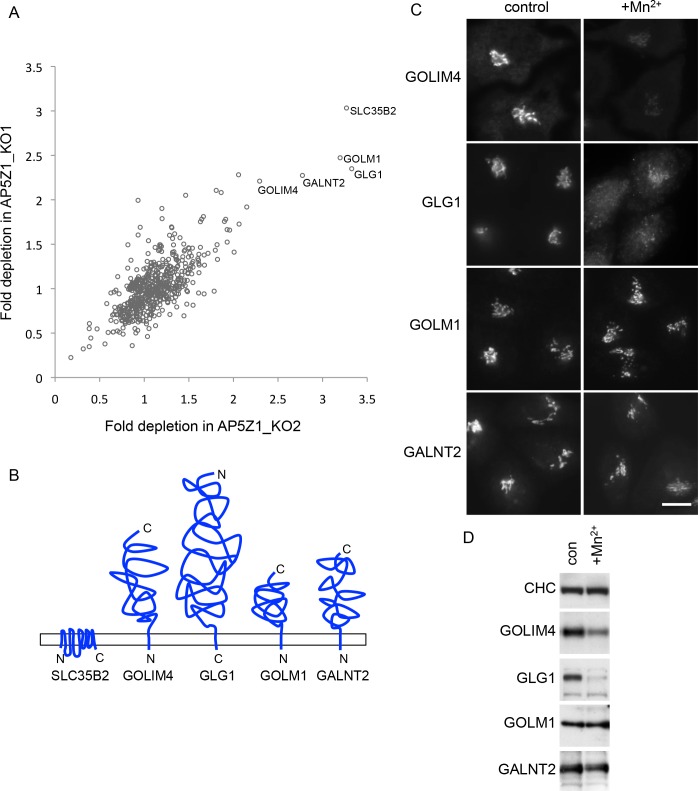
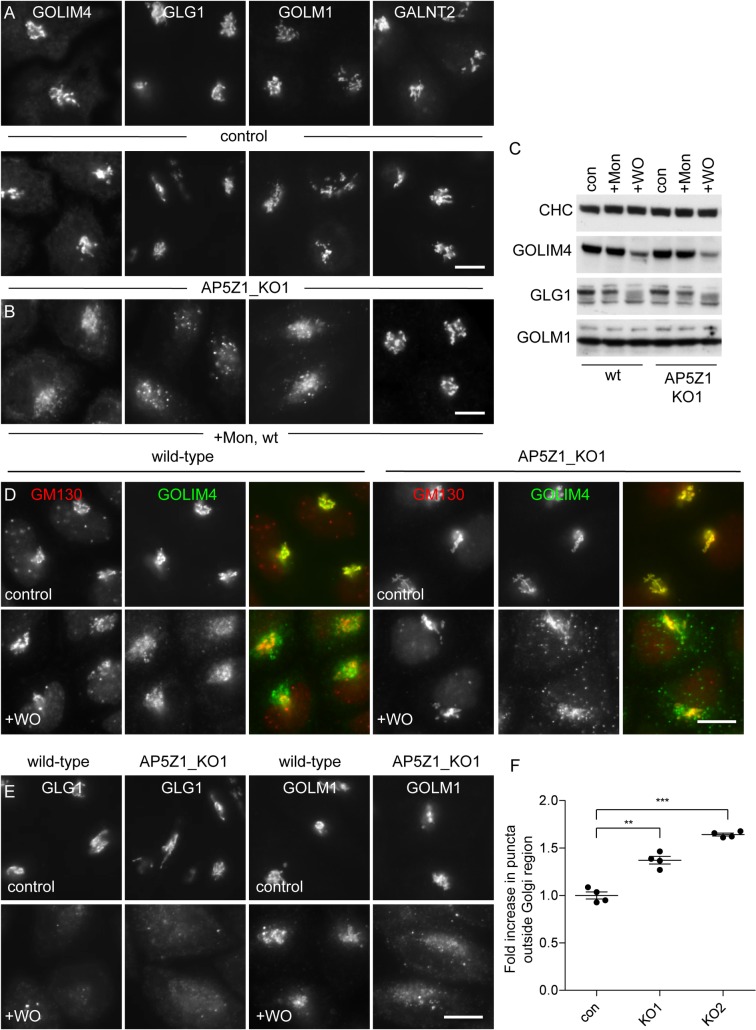
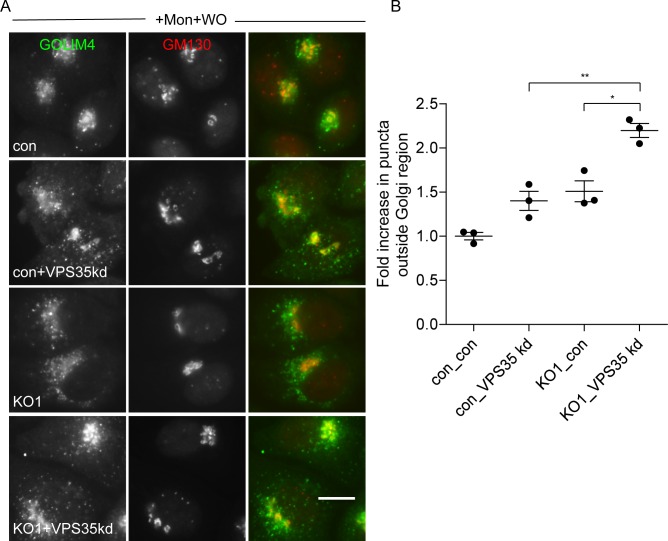
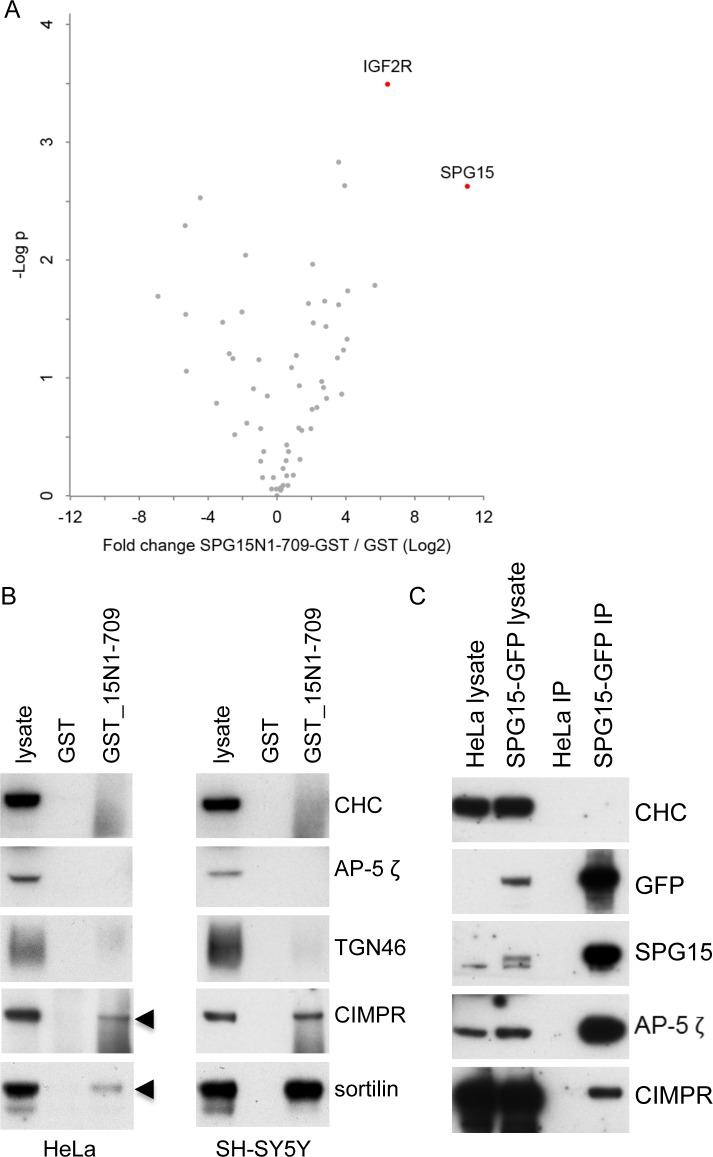
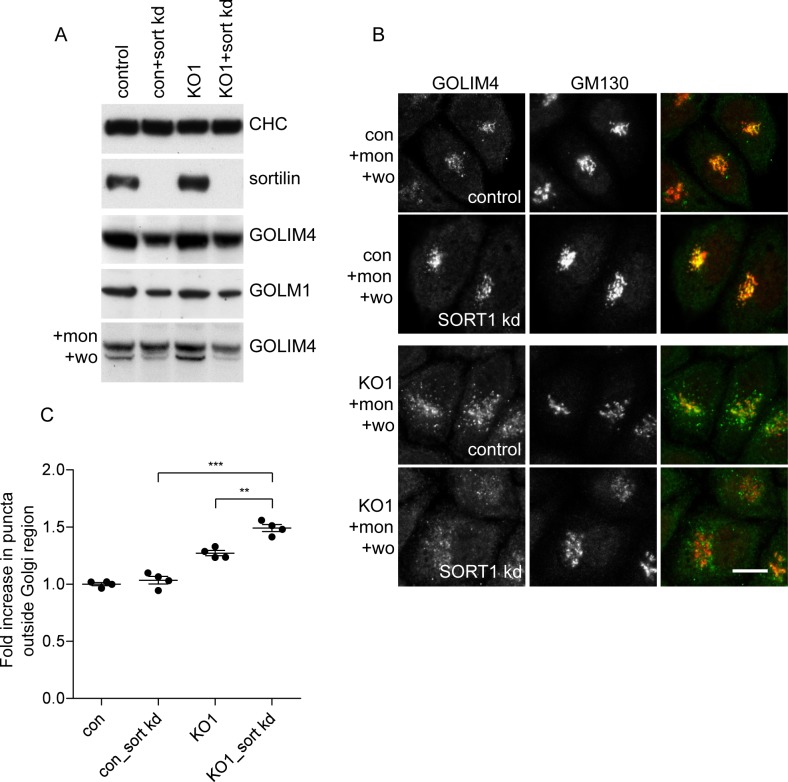
The AP-5 adaptor protein complex is presumed to function in membrane traffic, but so far nothing is known about its pathway or its cargo. We have used CRISPR-Cas9 to knock out the AP-5 ζ subunit gene, AP5Z1, in HeLa cells, and then analysed the phenotype by subcellular fractionation profiling and quantitative mass spectrometry. The retromer complex had an altered steady-state distribution in the knockout cells, and several Golgi proteins, including GOLIM4 and GOLM1, were depleted from vesicle-enriched fractions. Immunolocalisation showed that loss of AP-5 led to impaired retrieval of the cation-independent mannose 6-phosphate receptor (CIMPR), GOLIM4, and GOLM1 from endosomes back to the Golgi region. Knocking down the retromer complex exacerbated this phenotype. Both the CIMPR and sortilin interacted with the AP-5-associated protein SPG15 in pull-down assays, and we propose that sortilin may act as a link between Golgi proteins and the AP-5/SPG11/SPG15 complex. Together, our findings suggest that AP-5 functions in a novel sorting step out of late endosomes, acting as a backup pathway for retromer. This provides a mechanistic explanation for why mutations in AP-5/SPG11/SPG15 cause cells to accumulate aberrant endolysosomes, and highlights the role of endosome/lysosome dysfunction in the pathology of hereditary spastic paraplegia and other neurodegenerative disorders.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis.Autophagy. 2019 Jan;15(1):34-57. doi: 10.1080/15548627.2018.1507438. Epub 2018 Sep 13. Autophagy. 2019. PMID: 30081747 Free PMC article.
-
Identification of a novel conserved sorting motif required for retromer-mediated endosome-to-TGN retrieval.J Cell Sci. 2007 Jul 15;120(Pt 14):2378-89. doi: 10.1242/jcs.009654. J Cell Sci. 2007. PMID: 17606993
-
Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15.Mol Biol Cell. 2013 Aug;24(16):2558-69. doi: 10.1091/mbc.E13-03-0170. Epub 2013 Jul 3. Mol Biol Cell. 2013. PMID: 23825025 Free PMC article.
-
Retromer and the cation-independent mannose 6-phosphate receptor-Time for a trial separation?Traffic. 2018 Feb;19(2):150-152. doi: 10.1111/tra.12542. Epub 2017 Dec 21. Traffic. 2018. PMID: 29135085 Free PMC article. Review.
-
The role of AP-4 in cargo export from the trans-Golgi network and hereditary spastic paraplegia.Biochem Soc Trans. 2020 Oct 30;48(5):1877-1888. doi: 10.1042/BST20190664. Biochem Soc Trans. 2020. PMID: 33084855 Review.
Cited by
-
Retrograde transport of CDMPR depends on several machineries as analyzed by sulfatable nanobodies.Life Sci Alliance. 2022 Mar 21;5(7):e202101269. doi: 10.26508/lsa.202101269. Print 2022 Mar. Life Sci Alliance. 2022. PMID: 35314489 Free PMC article.
-
Assessing sub-cellular resolution in spatial proteomics experiments.Curr Opin Chem Biol. 2019 Feb;48:123-149. doi: 10.1016/j.cbpa.2018.11.015. Epub 2018 Dec 14. Curr Opin Chem Biol. 2019. PMID: 30711721 Free PMC article. Review.
-
Insights into Clinical, Genetic, and Pathological Aspects of Hereditary Spastic Paraplegias: A Comprehensive Overview.Front Mol Biosci. 2021 Nov 26;8:690899. doi: 10.3389/fmolb.2021.690899. eCollection 2021. Front Mol Biosci. 2021. PMID: 34901147 Free PMC article. Review.
-
Hereditary spastic paraplegia (SPG 48) with deafness and azoospermia: A case report.Front Neurol. 2023 Apr 3;14:1156100. doi: 10.3389/fneur.2023.1156100. eCollection 2023. Front Neurol. 2023. PMID: 37077568 Free PMC article.
-
Neuronal functions of adaptor complexes involved in protein sorting.Curr Opin Neurobiol. 2018 Aug;51:103-110. doi: 10.1016/j.conb.2018.02.021. Epub 2018 Mar 17. Curr Opin Neurobiol. 2018. PMID: 29558740 Free PMC article. Review.
References
-
- Robinson MS (2015) Forty years of clathrin-coated vesicles. Traffic 16: 1210–1238. doi: 10.1111/tra.12335 - DOI - PubMed
-
- Hirst J, Barlow LD, Francisco GC, Sahlender DA, Seaman MNJ, et al. (2011) The fifth adaptor protein complex. PLoS Biol 9: e1001170 doi: 10.1371/journal.pbio.1001170 - DOI - PMC - PubMed
-
- Hirst J, Borner GHH, Edgar J, Hein MY, Mann M, et al. (2013) Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell 24: 2558–2569. doi: 10.1091/mbc.E13-03-0170 - DOI - PMC - PubMed
-
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, et al. (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723. doi: 10.1016/j.cell.2015.09.053 - DOI - PubMed
-
- Itzhak DN, Tyanova S, Cox J, Borner GHH (2016) Global, quantitative and dynamic mapping of protein subcellular localization. eLife 5: e16950 doi: 10.7554/eLife.16950 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
