

Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer
- PMID: 28724614
- PMCID: PMC5558925
- DOI: 10.1101/gad.299958.117
Nuclear mTOR acts as a transcriptional integrator of the androgen signaling pathway in prostate cancer
Abstract
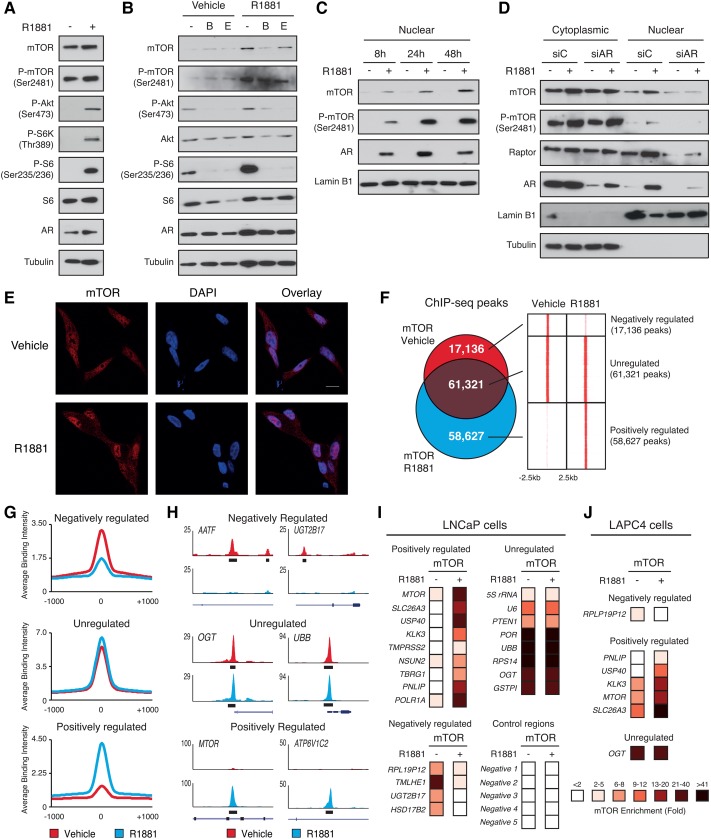
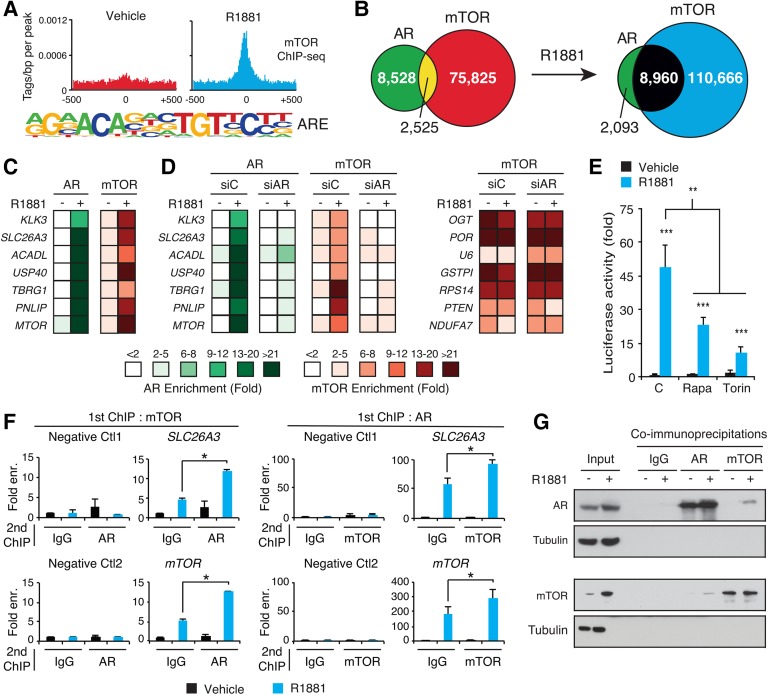
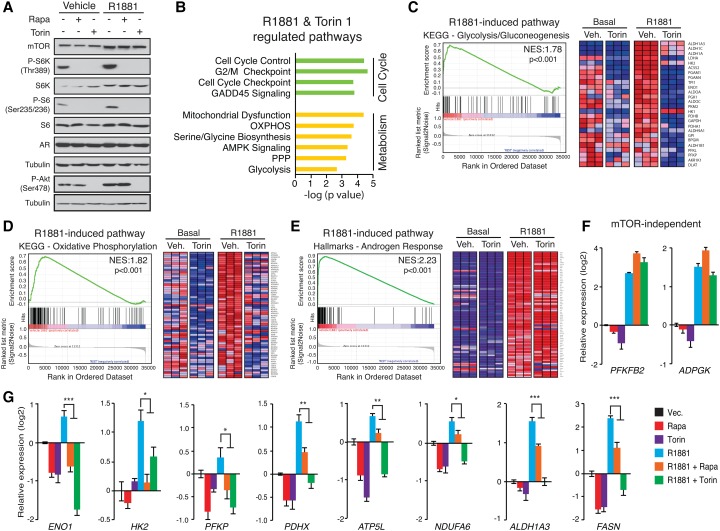
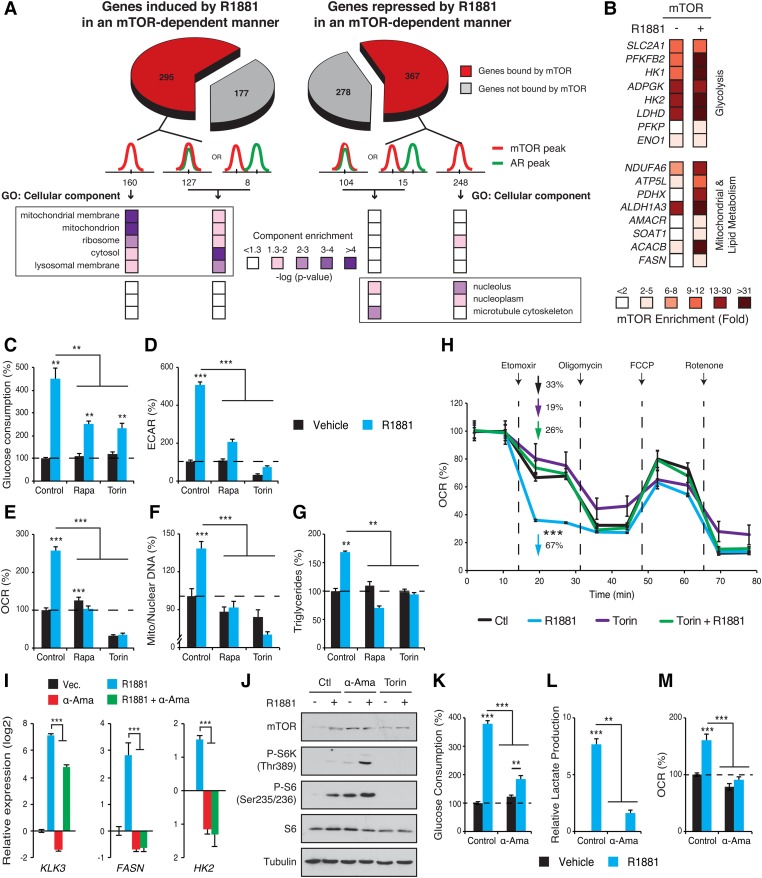
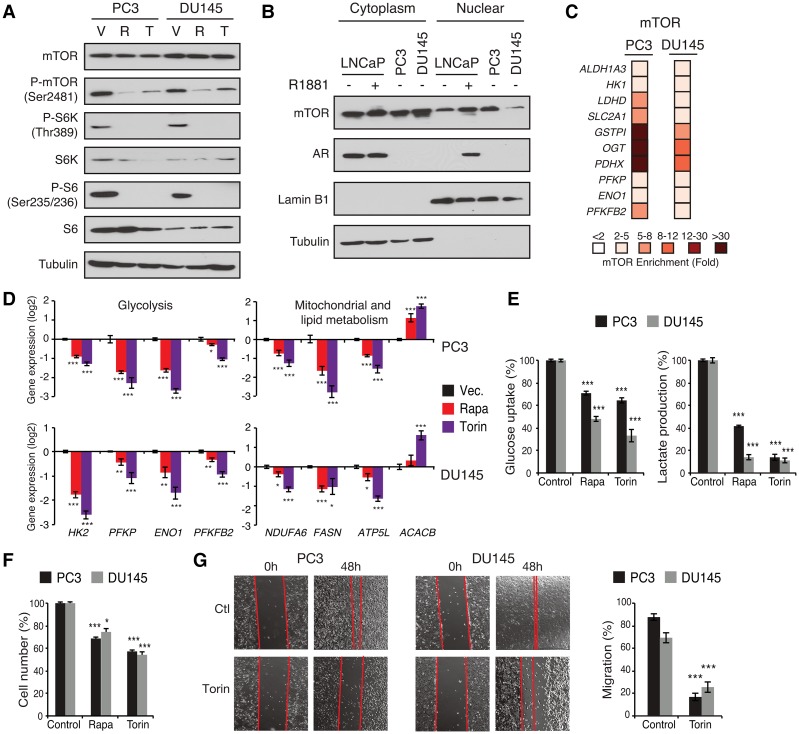
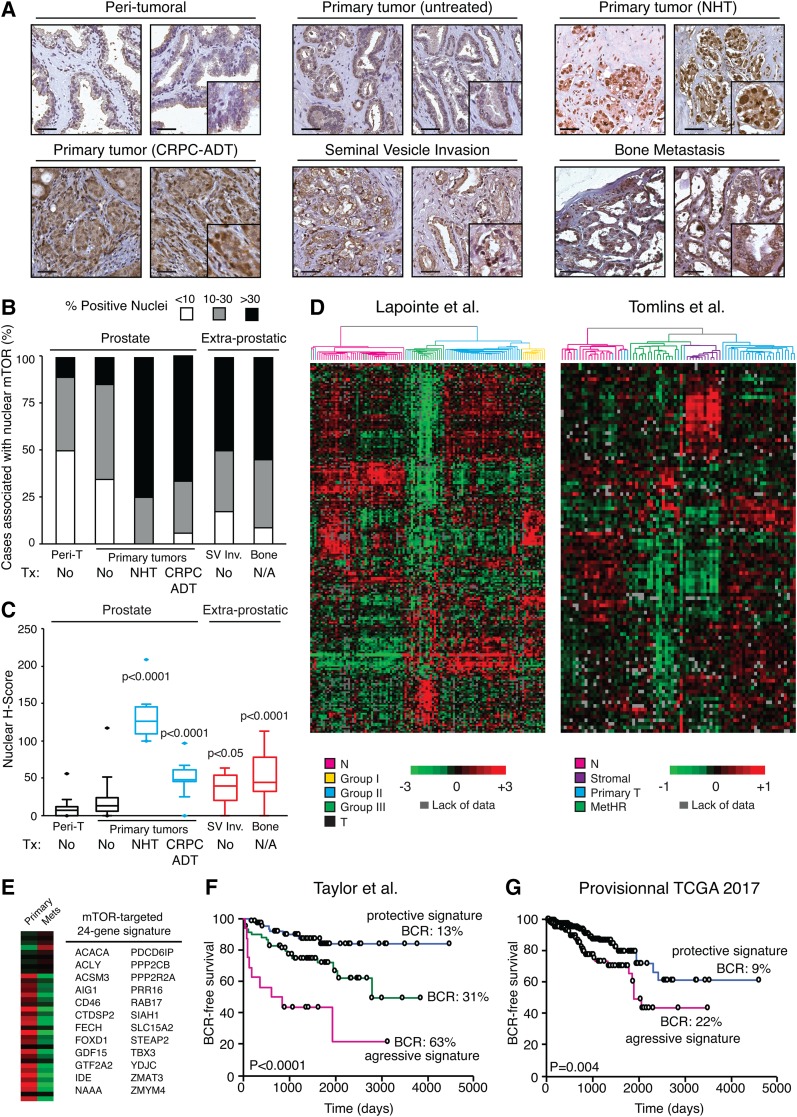
Androgen receptor (AR) signaling reprograms cellular metabolism to support prostate cancer (PCa) growth and survival. Another key regulator of cellular metabolism is mTOR, a kinase found in diverse protein complexes and cellular localizations, including the nucleus. However, whether nuclear mTOR plays a role in PCa progression and participates in direct transcriptional cross-talk with the AR is unknown. Here, via the intersection of gene expression, genomic, and metabolic studies, we reveal the existence of a nuclear mTOR-AR transcriptional axis integral to the metabolic rewiring of PCa cells. Androgens reprogram mTOR-chromatin associations in an AR-dependent manner in which activation of mTOR-dependent metabolic gene networks is essential for androgen-induced aerobic glycolysis and mitochondrial respiration. In models of castration-resistant PCa cells, mTOR was capable of transcriptionally regulating metabolic gene programs in the absence of androgens, highlighting a potential novel castration resistance mechanism to sustain cell metabolism even without a functional AR. Remarkably, we demonstrate that increased mTOR nuclear localization is indicative of poor prognosis in patients, with the highest levels detected in castration-resistant PCa tumors and metastases. Identification of a functional mTOR targeted multigene signature robustly discriminates between normal prostate tissues, primary tumors, and hormone refractory metastatic samples but is also predictive of cancer recurrence. This study thus underscores a paradigm shift from AR to nuclear mTOR as being the master transcriptional regulator of metabolism in PCa.
Keywords: CRPC; ChIP-seq; androgen receptor; energy metabolism; nuclear receptor; steroid.
© 2017 Audet-Walsh et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Canonical and Nuclear mTOR Specify Distinct Transcriptional Programs in Androgen-Dependent Prostate Cancer Cells.Mol Cancer Res. 2024 Feb 1;22(2):113-124. doi: 10.1158/1541-7786.MCR-23-0087. Mol Cancer Res. 2024. PMID: 37889103
-
SREBF1 Activity Is Regulated by an AR/mTOR Nuclear Axis in Prostate Cancer.Mol Cancer Res. 2018 Sep;16(9):1396-1405. doi: 10.1158/1541-7786.MCR-17-0410. Epub 2018 May 21. Mol Cancer Res. 2018. PMID: 29784665
-
Androgen receptors in hormone-dependent and castration-resistant prostate cancer.Pharmacol Ther. 2013 Dec;140(3):223-38. doi: 10.1016/j.pharmthera.2013.07.003. Epub 2013 Jul 13. Pharmacol Ther. 2013. PMID: 23859952 Review.
-
Androgen action in the prostate gland.Minerva Urol Nefrol. 2012 Mar;64(1):35-49. Minerva Urol Nefrol. 2012. PMID: 22402316 Review.
-
DNA-PK, Nuclear mTOR, and the Androgen Pathway in Prostate Cancer.Trends Cancer. 2020 Apr;6(4):337-347. doi: 10.1016/j.trecan.2020.01.015. Epub 2020 Feb 18. Trends Cancer. 2020. PMID: 32209447 Review.
Cited by
-
Systematic Evaluation for the Influences of the SOX17/Notch Receptor Family Members on Reversing Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cells.Front Oncol. 2021 Mar 10;11:607291. doi: 10.3389/fonc.2021.607291. eCollection 2021. Front Oncol. 2021. PMID: 33791203 Free PMC article.
-
The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling.Int J Mol Sci. 2020 Jun 25;21(12):4507. doi: 10.3390/ijms21124507. Int J Mol Sci. 2020. PMID: 32630372 Free PMC article. Review.
-
Reprogramming Carbohydrate Metabolism in Cancer and Its Role in Regulating the Tumor Microenvironment.Subcell Biochem. 2022;100:3-65. doi: 10.1007/978-3-031-07634-3_1. Subcell Biochem. 2022. PMID: 36301490 Free PMC article.
-
Transcriptional and Epigenetic Regulation by the Mechanistic Target of Rapamycin Complex 1 Pathway.J Mol Biol. 2018 Dec 7;430(24):4874-4890. doi: 10.1016/j.jmb.2018.10.008. Epub 2018 Oct 23. J Mol Biol. 2018. PMID: 30359581 Free PMC article. Review.
-
Oncogenic kinases and perturbations in protein synthesis machinery and energetics in neoplasia.J Mol Endocrinol. 2019 Feb 1;62(2):R83-R103. doi: 10.1530/JME-18-0058. J Mol Endocrinol. 2019. PMID: 30072418 Free PMC article. Review.
References
-
- Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, Bitting RL, Hobbs C, Soleau CF, Beer TM, et al. 2017. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer 81: 228–236. - PMC - PubMed
-
- Audet-Walsh E, Papadopoli DJ, Gravel SP, Yee T, Bridon G, Caron M, Bourque G, Giguère V, St-Pierre J. 2016. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep 14: 920–931. - PubMed
-
- Audet-Walsh E, Yee T, McGuirk S, Vernier M, Ouellet C, St-Pierre J, Giguere V. 2017. Androgen-dependent repression of ERRγ reprograms metabolism in prostate cancer. Cancer Res 77: 378–389. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous