

Thrombotic Microangiopathy in Inverted Formin 2 - Mediated Renal Disease
- PMID: 27974406
- PMCID: PMC5373440
- DOI: 10.1681/ASN.2015101189
Thrombotic Microangiopathy in Inverted Formin 2 - Mediated Renal Disease
Abstract
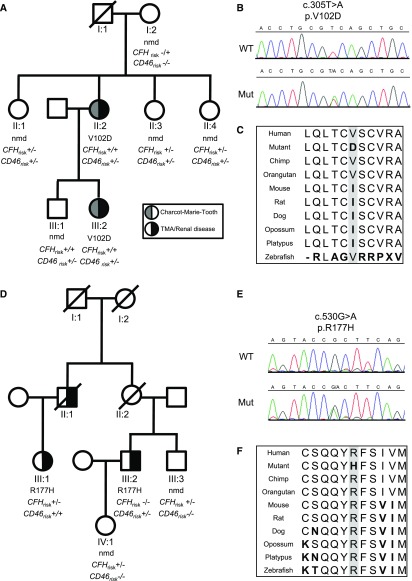
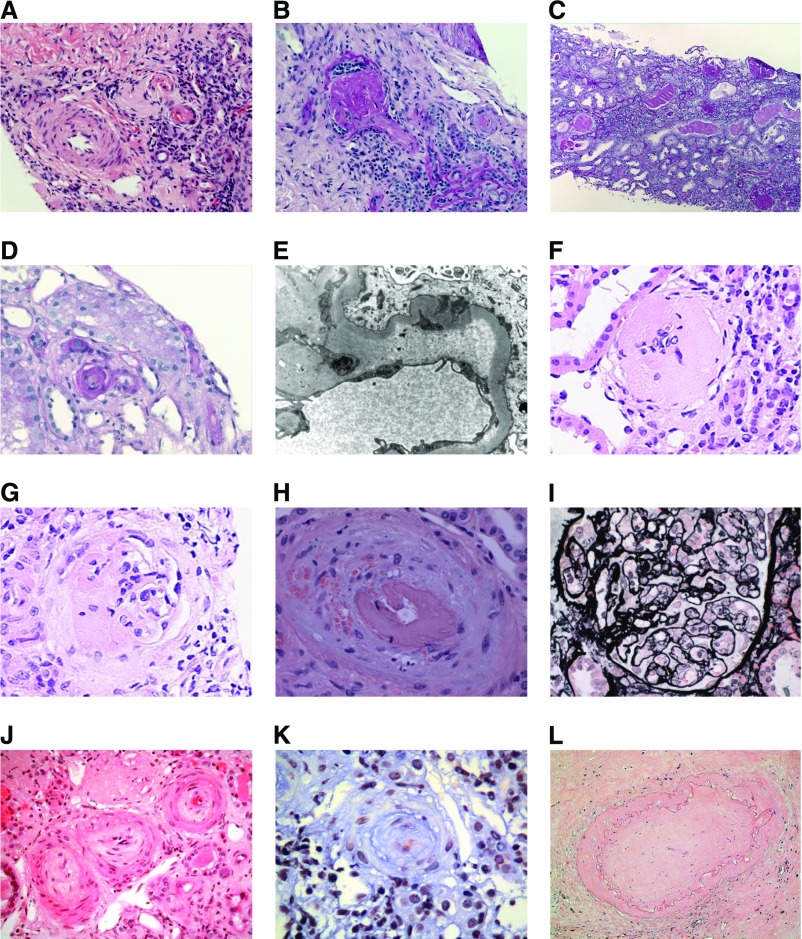
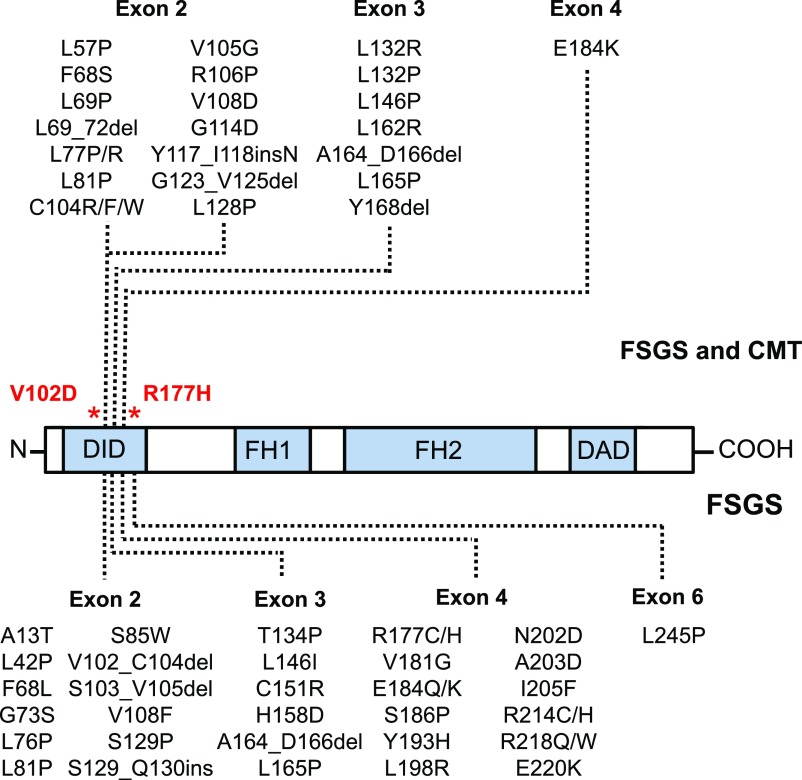
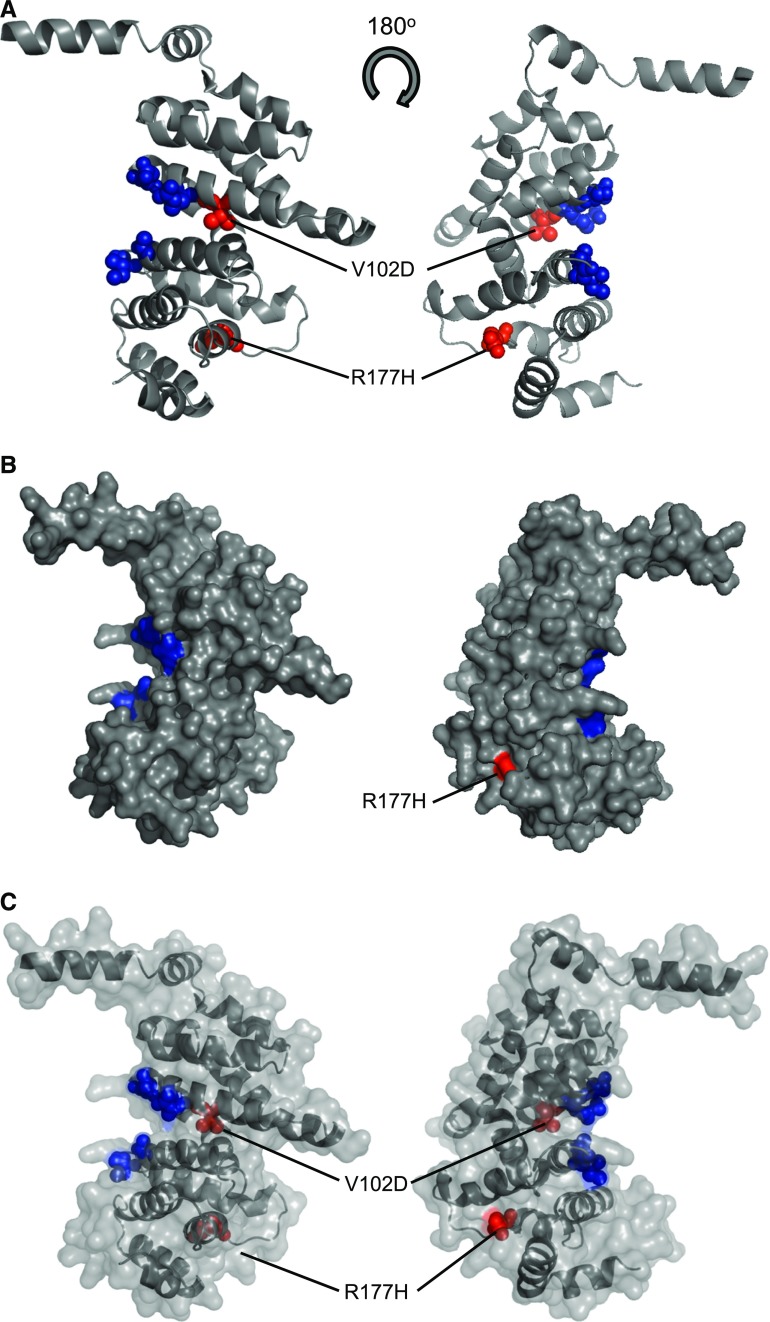
The demonstration of impaired C regulation in the thrombotic microangiopathy (TMA) atypical hemolytic uremic syndrome (aHUS) resulted in the successful introduction of the C inhibitor eculizumab into clinical practice. C abnormalities account for approximately 50% of aHUS cases; however, mutations in the non-C gene diacylglycerol kinase-ε have been described recently in individuals not responsive to eculizumab. We report here a family in which the proposita presented with aHUS but did not respond to eculizumab. Her mother had previously presented with a post-renal transplant TMA. Both the proposita and her mother also had Charcot-Marie-Tooth disease. Using whole-exome sequencing, we identified a mutation in the inverted formin 2 gene (INF2) in the mutational hotspot for FSGS. Subsequent analysis of the Newcastle aHUS cohort identified another family with a functionally-significant mutation in INF2 In this family, renal transplantation was associated with post-transplant TMA. All individuals with INF2 mutations presenting with a TMA also had aHUS risk haplotypes, potentially accounting for the genetic pleiotropy. Identifying individuals with TMAs who may not respond to eculizumab will avoid prolonged exposure of such individuals to the infectious complications of terminal pathway C blockade.
Keywords: complement; focal segmental glomerulosclerosis; hemolytic uremic syndrome.
Copyright © 2017 by the American Society of Nephrology.
Figures
Similar articles
-
Isolated thrombotic microangiopathy of the small intestine in a patient with atypical hemolytic uremic syndrome - a case report.BMC Nephrol. 2020 Mar 24;21(1):104. doi: 10.1186/s12882-020-01766-0. BMC Nephrol. 2020. PMID: 32204691 Free PMC article.
-
Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study.BMC Nephrol. 2019 Apr 10;20(1):125. doi: 10.1186/s12882-019-1314-1. BMC Nephrol. 2019. PMID: 30971227 Free PMC article.
-
A case-based narrative review of pregnancy-associated atypical hemolytic uremic syndrome/complement-mediated thrombotic microangiopathy.Kidney Int. 2024 May;105(5):960-970. doi: 10.1016/j.kint.2023.12.021. Epub 2024 Feb 24. Kidney Int. 2024. PMID: 38408703
-
Thrombotic microangiopathy in aHUS and beyond: clinical clues from complement genetics.Nat Rev Nephrol. 2021 Aug;17(8):543-553. doi: 10.1038/s41581-021-00424-4. Epub 2021 May 5. Nat Rev Nephrol. 2021. PMID: 33953366 Review.
-
An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document.Nefrologia. 2015;35(5):421-47. doi: 10.1016/j.nefro.2015.07.005. Epub 2015 Oct 9. Nefrologia. 2015. PMID: 26456110 English, Spanish.
Cited by
-
Adeno-associated virus-based gene therapy treats inflammatory kidney disease in mice.J Clin Invest. 2024 Aug 15;134(17):e174722. doi: 10.1172/JCI174722. J Clin Invest. 2024. PMID: 39225099 Free PMC article.
-
Dysregulation of Immune Cell Subpopulations in Atypical Hemolytic Uremic Syndrome.Int J Mol Sci. 2023 Jun 11;24(12):10007. doi: 10.3390/ijms241210007. Int J Mol Sci. 2023. PMID: 37373158 Free PMC article.
-
Atypical hemolytic uremic syndrome in the era of terminal complement inhibition: an observational cohort study.Blood. 2023 Oct 19;142(16):1371-1386. doi: 10.1182/blood.2022018833. Blood. 2023. PMID: 37369098 Free PMC article.
-
Variants in complement genes are uncommon in patients with anti-factor H autoantibody-associated atypical hemolytic uremic syndrome.Pediatr Nephrol. 2023 Aug;38(8):2659-2668. doi: 10.1007/s00467-022-05862-1. Epub 2023 Jan 9. Pediatr Nephrol. 2023. PMID: 36622444
-
Diagnosis and treatment of thrombotic microangiopathy.Int J Lab Hematol. 2022 Sep;44 Suppl 1(Suppl 1):101-113. doi: 10.1111/ijlh.13954. Int J Lab Hematol. 2022. PMID: 36074708 Free PMC article. Review.
References
-
- Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, Bingham C, Cohen DJ, Delmas Y, Douglas K, Eitner F, Feldkamp T, Fouque D, Furman RR, Gaber O, Herthelius M, Hourmant M, Karpman D, Lebranchu Y, Mariat C, Menne J, Moulin B, Nürnberger J, Ogawa M, Remuzzi G, Richard T, Sberro-Soussan R, Severino B, Sheerin NS, Trivelli A, Zimmerhackl LB, Goodship T, Loirat C: Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368: 2169–2181, 2013 - PubMed
-
- Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, Ji W, Overton JD, Mane SM, Nürnberg G, Altmüller J, Thiele H, Morin D, Deschenes G, Baudouin V, Llanas B, Collard L, Majid MA, Simkova E, Nürnberg P, Rioux-Leclerc N, Moeckel GW, Gubler MC, Hwa J, Loirat C, Lifton RP: Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45: 531–536, 2013 - PMC - PubMed
-
- Ozaltin F, Li B, Rauhauser A, An SW, Soylemezoglu O, Gonul II, Taskiran EZ, Ibsirlioglu T, Korkmaz E, Bilginer Y, Duzova A, Ozen S, Topaloglu R, Besbas N, Ashraf S, Du Y, Liang C, Chen P, Lu D, Vadnagara K, Arbuckle S, Lewis D, Wakeland B, Quigg RJ, Ransom RF, Wakeland EK, Topham MK, Bazan NG, Mohan C, Hildebrandt F, Bakkaloglu A, Huang CL, Attanasio M: DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol 24: 377–384, 2013 - PMC - PubMed
-
- Nishimura J, Yamamoto M, Hayashi S, Ohyashiki K, Ando K, Brodsky AL, Noji H, Kitamura K, Eto T, Takahashi T, Masuko M, Matsumoto T, Wano Y, Shichishima T, Shibayama H, Hase M, Li L, Johnson K, Lazarowski A, Tamburini P, Inazawa J, Kinoshita T, Kanakura Y: Genetic variants in C5 and poor response to eculizumab. N Engl J Med 370: 632–639, 2014 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
