

Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients
- PMID: 27283993
- PMCID: PMC4906406
- DOI: 10.1038/ncomms11815
Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients
Erratum in
-
Corrigendum: Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients.Nat Commun. 2016 Nov 14;7:13513. doi: 10.1038/ncomms13513. Nat Commun. 2016. PMID: 27841271 Free PMC article. No abstract available.
Abstract
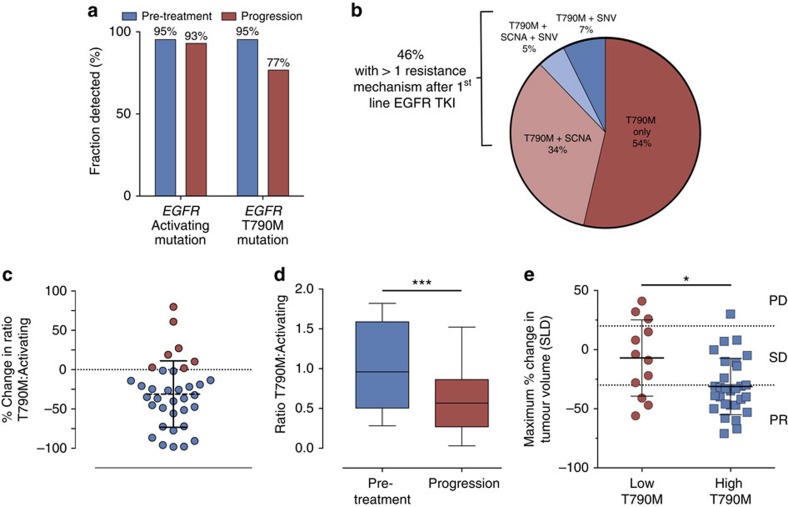
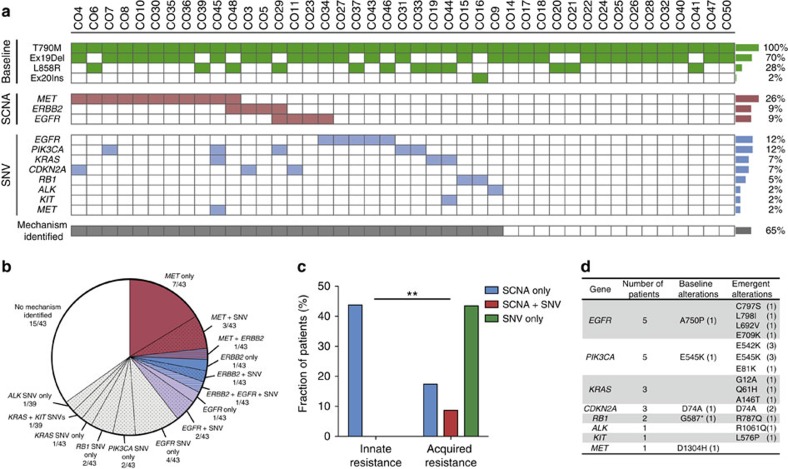
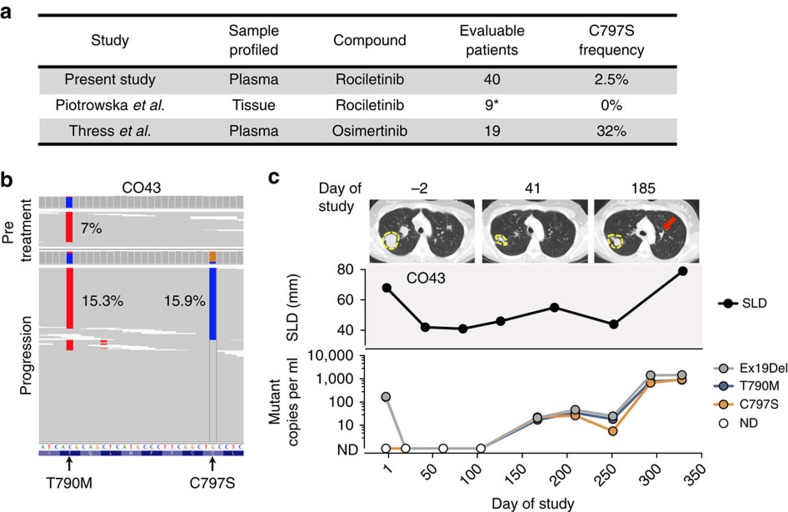
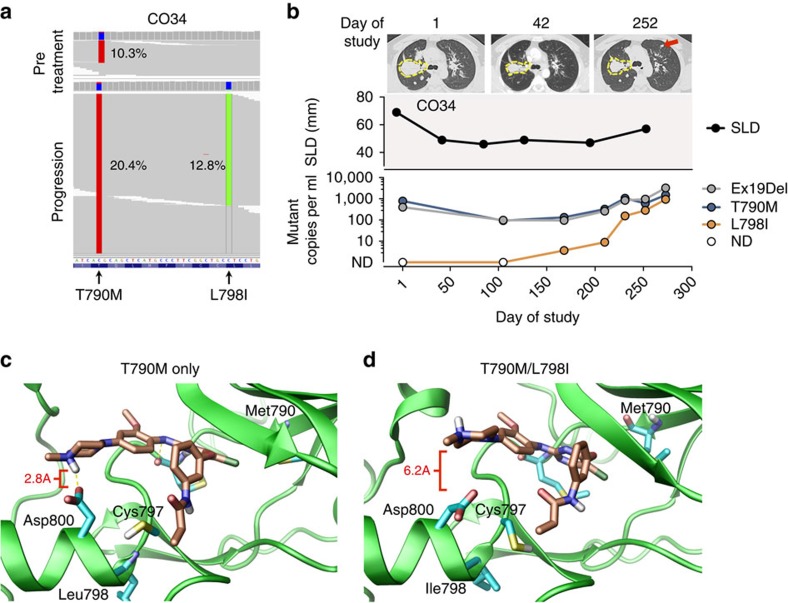
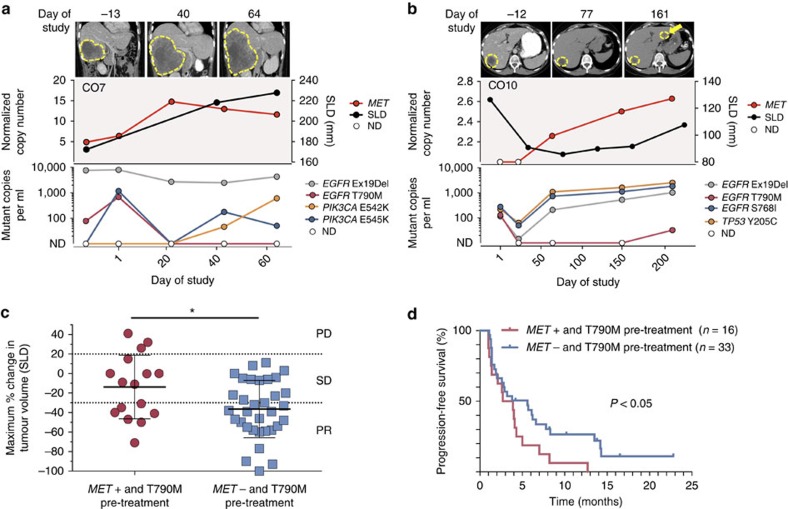
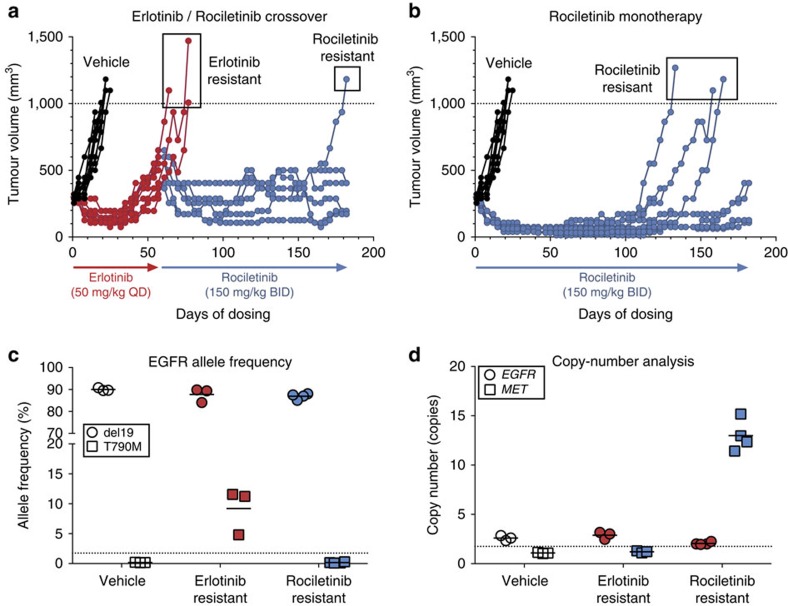
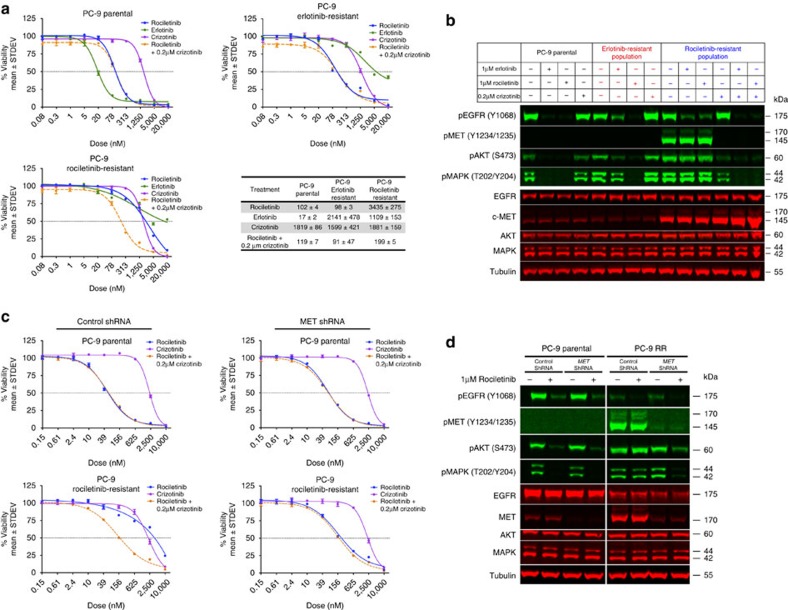
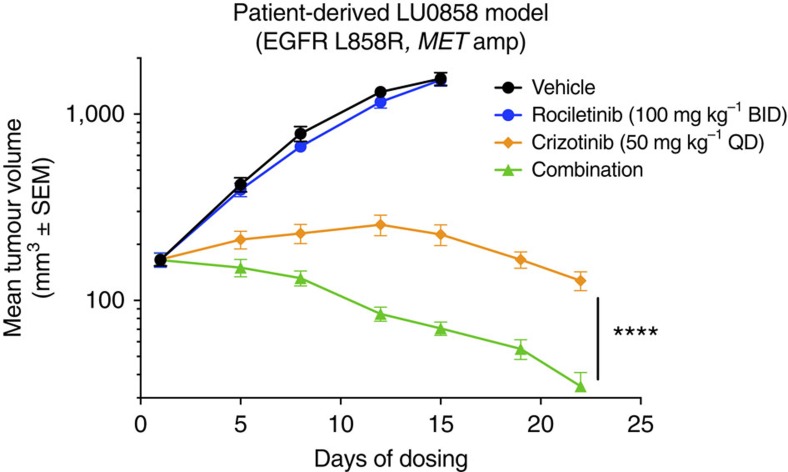
Circulating tumour DNA (ctDNA) analysis facilitates studies of tumour heterogeneity. Here we employ CAPP-Seq ctDNA analysis to study resistance mechanisms in 43 non-small cell lung cancer (NSCLC) patients treated with the third-generation epidermal growth factor receptor (EGFR) inhibitor rociletinib. We observe multiple resistance mechanisms in 46% of patients after treatment with first-line inhibitors, indicating frequent intra-patient heterogeneity. Rociletinib resistance recurrently involves MET, EGFR, PIK3CA, ERRB2, KRAS and RB1. We describe a novel EGFR L798I mutation and find that EGFR C797S, which arises in ∼33% of patients after osimertinib treatment, occurs in <3% after rociletinib. Increased MET copy number is the most frequent rociletinib resistance mechanism in this cohort and patients with multiple pre-existing mechanisms (T790M and MET) experience inferior responses. Similarly, rociletinib-resistant xenografts develop MET amplification that can be overcome with the MET inhibitor crizotinib. These results underscore the importance of tumour heterogeneity in NSCLC and the utility of ctDNA-based resistance mechanism assessment.
Conflict of interest statement
A.M.N., A.A.A. and M.D. are co-inventors on patent applications, ‘An analytical platform for patient-specific profiling of circulating neoplastic DNA' 14/209,807 and, ‘Practical Methods for Patient-Specific Tumour Markers for Disease Monitoring of Cancer' PCT/US2015/049838, related to CAPP-Seq. A.M.N., A.A.A. and M.D. are consultants for Roche Molecular Diagnostics. A.D.S., C.A.K., H.J.H. and T.C.H. are employees of Clovis Oncology. C.A.K. has ownership interest in Clovis Oncology. Z.P. has received speakers bureau honoraria from Clovis Oncology. H.A.W. has received commercial research grants from Clovis Oncology. L.V.S. and J.W.N. are consultants for Clovis Oncology. The remaining authors declare no competing financial interests.
Figures
Comment in
-
Circulating tumour DNA and resistance mechanisms during EGFR inhibitor therapy in lung cancer.J Thorac Dis. 2016 Sep;8(9):2357-2359. doi: 10.21037/jtd.2016.07.96. J Thorac Dis. 2016. PMID: 27746975 Free PMC article. No abstract available.
-
Liquid biopsy in the practice of neo-oncology.J Thorac Dis. 2016 Oct;8(10):E1279-E1281. doi: 10.21037/jtd.2016.10.72. J Thorac Dis. 2016. PMID: 27867607 Free PMC article. No abstract available.
Similar articles
-
Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment.Cancer Lett. 2016 Oct 1;380(2):494-504. doi: 10.1016/j.canlet.2016.07.021. Epub 2016 Jul 19. Cancer Lett. 2016. PMID: 27450722
-
Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors.PLoS One. 2013 Dec 26;8(12):e84700. doi: 10.1371/journal.pone.0084700. eCollection 2013. PLoS One. 2013. PMID: 24386407 Free PMC article.
-
Genetic Profiling of Non-Small Cell Lung Cancer at Development of Resistance to First- or Second-Generation EGFR-TKIs by CAPP-Seq Analysis of Circulating Tumor DNA.Oncologist. 2019 Aug;24(8):1022-1026. doi: 10.1634/theoncologist.2019-0101. Epub 2019 Apr 25. Oncologist. 2019. PMID: 31023862 Free PMC article.
-
Treatment of Brain Metastases of Non-Small Cell Lung Carcinoma.Int J Mol Sci. 2021 Jan 8;22(2):593. doi: 10.3390/ijms22020593. Int J Mol Sci. 2021. PMID: 33435596 Free PMC article. Review.
-
Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer.J Hematol Oncol. 2016 Apr 12;9:34. doi: 10.1186/s13045-016-0268-z. J Hematol Oncol. 2016. PMID: 27071706 Free PMC article. Review.
Cited by
-
CAPP-seq analysis of circulating tumor DNA from patients with EGFR T790M-positive lung cancer after osimertinib.Int J Clin Oncol. 2021 Sep;26(9):1628-1639. doi: 10.1007/s10147-021-01947-3. Epub 2021 Jun 11. Int J Clin Oncol. 2021. PMID: 34117553
-
erbB in NSCLC as a molecular target: current evidences and future directions.ESMO Open. 2020 Aug;5(4):e000724. doi: 10.1136/esmoopen-2020-000724. ESMO Open. 2020. PMID: 32820012 Free PMC article. Review.
-
Expanding anaplastic lymphoma kinase therapeutic indication to early stage non-small cell lung cancer.Transl Lung Cancer Res. 2019 Nov;8(Suppl 3):S290-S297. doi: 10.21037/tlcr.2019.07.07. Transl Lung Cancer Res. 2019. PMID: 31857952 Free PMC article. Review.
-
Clinical significance of repeat rebiopsy in detecting the EGFR T790M secondary mutation in patients with non-small cell lung cancer.Oncotarget. 2018 Jun 29;9(50):29525-29531. doi: 10.18632/oncotarget.25705. eCollection 2018 Jun 29. Oncotarget. 2018. PMID: 30034635 Free PMC article.
-
High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants.Nat Med. 2019 Dec;25(12):1928-1937. doi: 10.1038/s41591-019-0652-7. Epub 2019 Nov 25. Nat Med. 2019. PMID: 31768066 Free PMC article. Clinical Trial.
References
-
- Paez J. G. et al.. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004). - PubMed
-
- Lynch T. J. et al.. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139 (2004). - PubMed
-
- Mok T. S. et al.. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009). - PubMed
-
- Maemondo M. et al.. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388 (2010). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
