

Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases
- PMID: 27130805
- PMCID: PMC7112520
- DOI: 10.1016/j.pharmthera.2016.03.018
Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases
Abstract
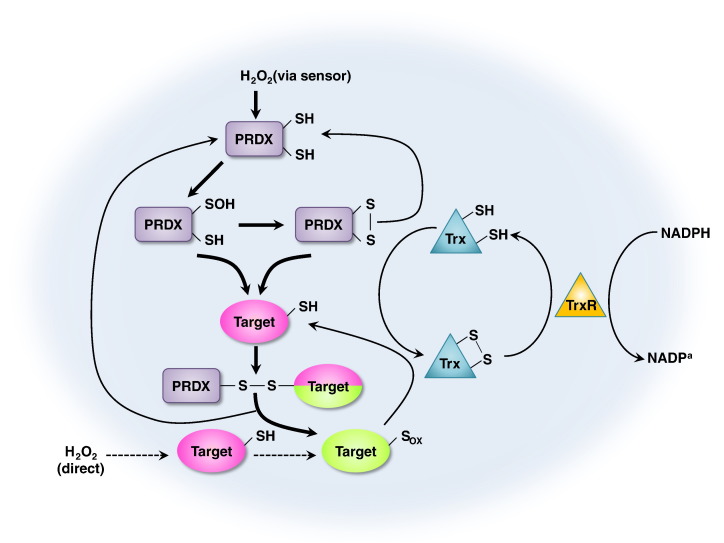
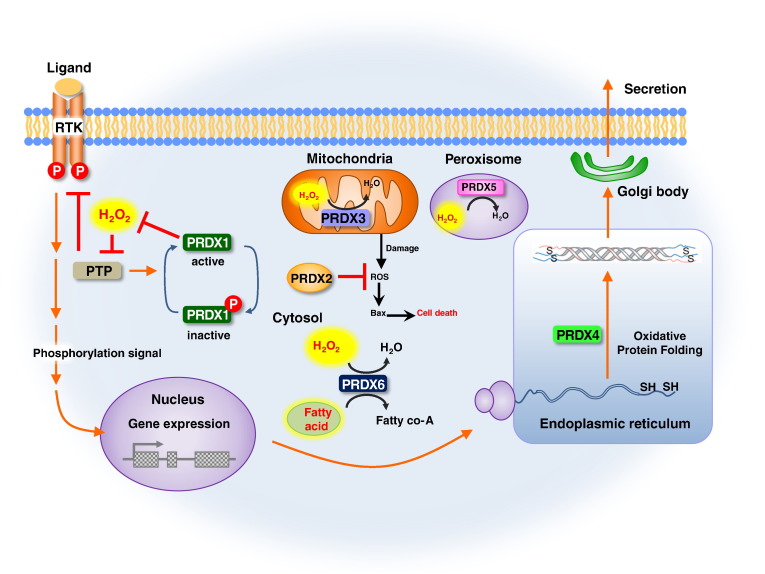
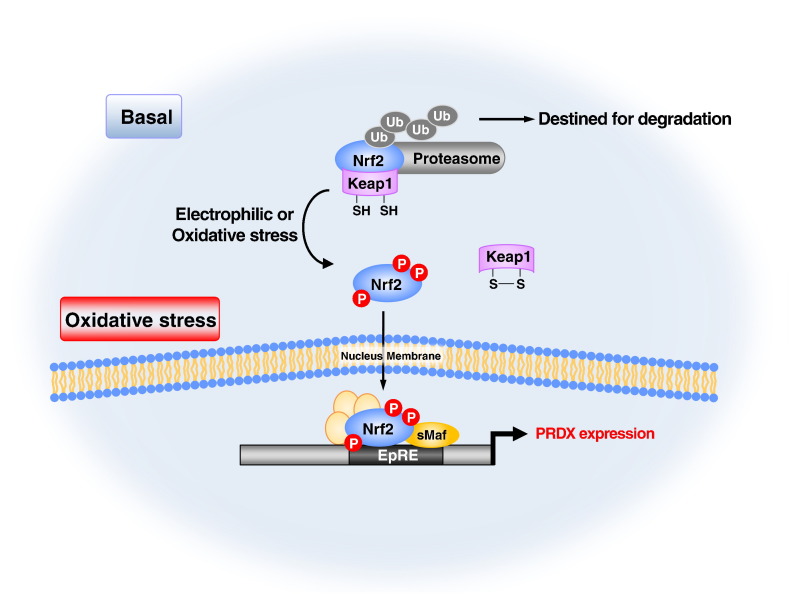
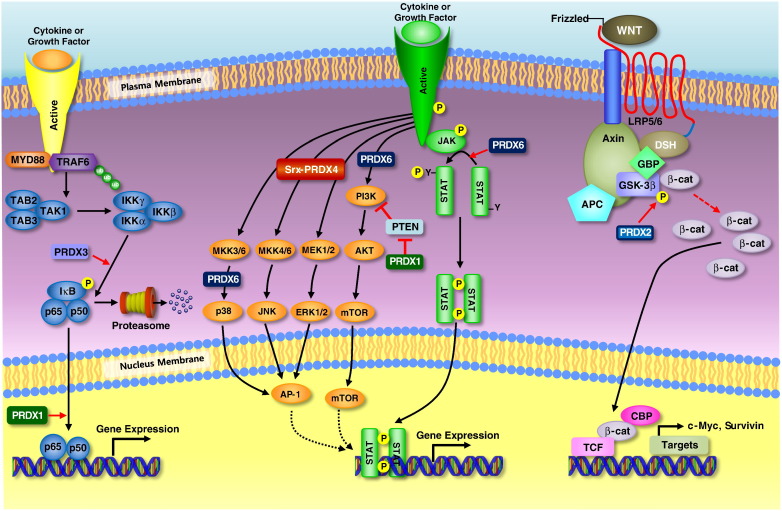
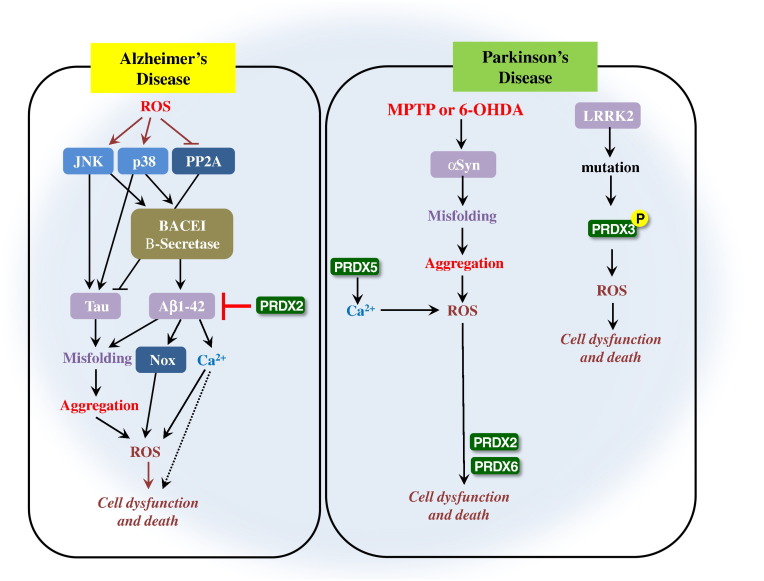
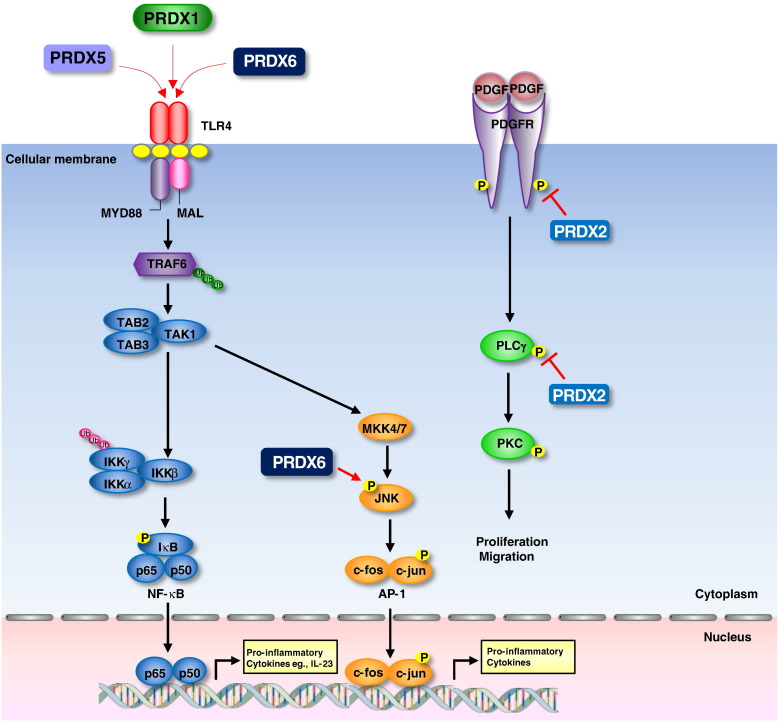
Peroxiredoxins (PRDXs) are antioxidant enzymes, known to catalyze peroxide reduction to balance cellular hydrogen peroxide (H2O2) levels, which are essential for cell signaling and metabolism and act as a regulator of redox signaling. Redox signaling is a critical component of cell signaling pathways that are involved in the regulation of cell growth, metabolism, hormone signaling, immune regulation and variety of other physiological functions. Early studies demonstrated that PRDXs regulates cell growth, metabolism and immune regulation and therefore involved in the pathologic regulator or protectant of several cancers, neurodegenerative diseases and inflammatory diseases. Oxidative stress and antioxidant systems are important regulators of redox signaling regulated diseases. In addition, thiol-based redox systems through peroxiredoxins have been demonstrated to regulate several redox-dependent process related diseases. In this review article, we will discuss recent findings regarding PRDXs in the development of diseases and further discuss therapeutic approaches targeting PRDXs. Moreover, we will suggest that PRDXs could be targets of several diseases and the therapeutic agents for targeting PRDXs may have potential beneficial effects for the treatment of cancers, neurodegenerative diseases and inflammatory diseases. Future research should open new avenues for the design of novel therapeutic approaches targeting PRDXs.
Keywords: Cancer; Inflammatory diseases; Neurodegenerative diseases; Peroxiredoxins; Therapeutic approaches.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Functions of Peroxiredoxins and Their Roles in Autoimmune Diseases.Antioxid Redox Signal. 2024 Feb;40(4-6):329-344. doi: 10.1089/ars.2022.0139. Epub 2023 Mar 27. Antioxid Redox Signal. 2024. PMID: 36738225 Review.
-
Peroxiredoxin 1 and its role in cell signaling.Cell Cycle. 2009 Dec 15;8(24):4072-8. doi: 10.4161/cc.8.24.10242. Epub 2009 Dec 5. Cell Cycle. 2009. PMID: 19923889 Free PMC article. Review.
-
New insights into the roles of peroxiredoxins in cancer.Biomed Pharmacother. 2023 Aug;164:114896. doi: 10.1016/j.biopha.2023.114896. Epub 2023 May 19. Biomed Pharmacother. 2023. PMID: 37210897 Review.
-
Peroxiredoxins and Beyond; Redox Systems Regulating Lung Physiology and Disease.Antioxid Redox Signal. 2019 Nov 10;31(14):1070-1091. doi: 10.1089/ars.2019.7752. Epub 2019 Apr 5. Antioxid Redox Signal. 2019. PMID: 30799628 Free PMC article. Review.
-
The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs.Antioxid Redox Signal. 2018 Mar 1;28(7):558-573. doi: 10.1089/ars.2017.7162. Epub 2017 Jul 17. Antioxid Redox Signal. 2018. PMID: 28587525 Review.
Cited by
-
Foot-and-Mouth Disease Virus: Molecular Interplays with IFN Response and the Importance of the Model.Viruses. 2022 Sep 27;14(10):2129. doi: 10.3390/v14102129. Viruses. 2022. PMID: 36298684 Free PMC article. Review.
-
The Effect of Neurotoxin MPTP and Neuroprotector Isatin on the Profile of Ubiquitinated Brain Mitochondrial Proteins.Cells. 2018 Jul 31;7(8):91. doi: 10.3390/cells7080091. Cells. 2018. PMID: 30065189 Free PMC article.
-
Peroxiredoxins as Markers of Oxidative Stress in IgA Nephropathy, Membranous Nephropathy and Lupus Nephritis.Arch Immunol Ther Exp (Warsz). 2021 Dec 16;70(1):3. doi: 10.1007/s00005-021-00638-1. Arch Immunol Ther Exp (Warsz). 2021. PMID: 34914001 Free PMC article.
-
Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins.J Biol Chem. 2018 Jul 27;293(30):11901-11912. doi: 10.1074/jbc.RA117.001690. Epub 2018 Jun 8. J Biol Chem. 2018. PMID: 29884768 Free PMC article.
-
Determination of Highly Sensitive Biological Cell Model Systems to Screen BPA-Related Health Hazards Using Pathway Studio.Int J Mol Sci. 2017 Sep 6;18(9):1909. doi: 10.3390/ijms18091909. Int J Mol Sci. 2017. PMID: 28878155 Free PMC article.
References
-
- Abbas K., Breton J., Picot C.R., Quesniaux V., Bouton C., Drapier J.C. Signaling events leading to peroxiredoxin 5 up-regulation in immunostimulated macrophages. Free Radic Biol Med. 2009;47:794–802. - PubMed
-
- Adler V., Yin Z., Tew K.D., Ronai Z. Role of redox potential and reactive oxygen species in stress signaling. Oncogene. 1999;18:6104–6111. - PubMed
-
- Agrawal Singh S., Isken F., Agelopoulos K., Klein H.U., Thoennissen N.H., Koehler G. Genome-wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood. 2012;119:2346–2357. - PubMed
-
- Angeles D.C., Gan B.H., Onstead L., Zhao Y., Lim K.L., Dachsel J. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum Mutat. 2011;32:1390–1397. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
