

The P42 peptide and Peptide-based therapies for Huntington's disease
- PMID: 26984770
- PMCID: PMC4794846
- DOI: 10.1186/s13023-016-0405-3
The P42 peptide and Peptide-based therapies for Huntington's disease
Abstract
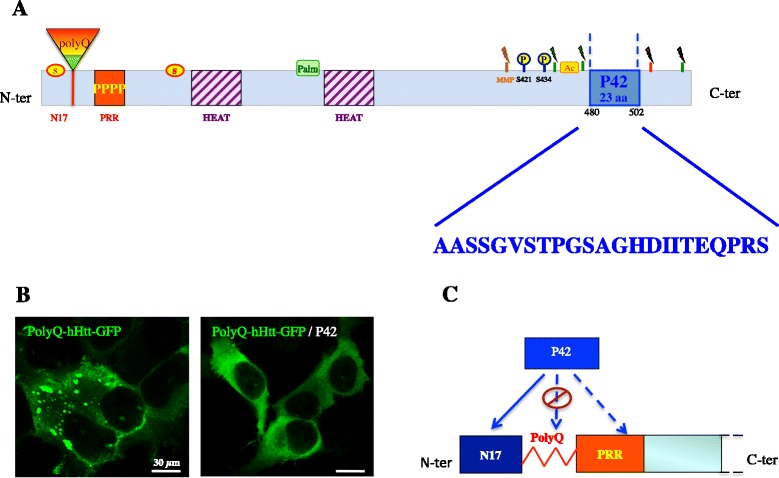
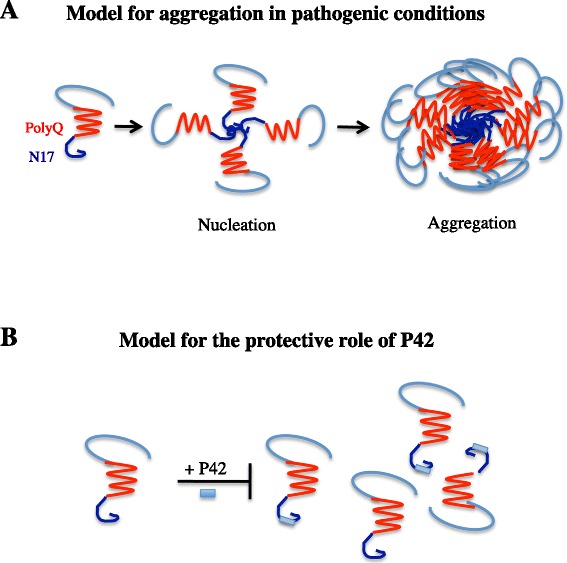
Huntington's disease (HD) is a progressive neurodegenerative hereditary disease clinically characterised by the presence of involuntary movements, behavioural problems and cognitive decline. The disease-onset is usually between 30 and 50 years of age. HD is a rare disorder affecting approximately 1.3 in 10,000 people in the European Union. It is caused by an expanded CAG repeat in the first exon of the Huntingtin (HTT) gene, leading to an abnormal form of the Huntingtin protein (Htt) (polyQHtt), containing N-terminus, enlarged polyglutamine strands of variable length that stick together to form aggregates and nuclear inclusions in the damaged brain cells. Treatments currently used for Huntington's disease are symptomatic and aimed at temporally relieving the symptoms of the disease; although some promising therapies are on study, there is no drug capable of stopping disease progression either in the form of delaying onset or slowing disability progression. The utilization of peptides interacting with polyQ stretches or with Htt protein to prevent misfolding and aggregation of the expanded polyQ protein is a fascinating idea, because of low potential toxicity and ability to target very initial steps in the pathophysiological cascade of the disease, such as aggregation or cleavage process. Indeed, several therapeutic peptides have been developed and were found to significantly slow down the progression of symptoms in experimental models of Huntington's disease. This review is essentially focusing on the latest development concerning peptide strategy. In particular, we focused on a 23aa peptide P42, which is a part of the Htt protein. It is expected to work principally by preventing the abnormal Htt protein from sticking together, thereby preventing pathological consequences of aggregation and improving the symptoms of the disease. In the meantime, as P42 is part of the Htt protein, some therapeutic properties might be linked to the physiological actions of the peptide itself, considered as a functional domain of the Htt protein.
Keywords: Huntington’s disease; P42; Peptide-based therapy.
Figures
Similar articles
-
In silico designing of putative peptides for targeting pathological protein Htt in Huntington's disease.Heliyon. 2021 Feb 12;7(2):e06088. doi: 10.1016/j.heliyon.2021.e06088. eCollection 2021 Feb. Heliyon. 2021. PMID: 33659724 Free PMC article. Review.
-
Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models.Hum Mol Genet. 2006 Sep 15;15(18):2743-51. doi: 10.1093/hmg/ddl210. Epub 2006 Aug 7. Hum Mol Genet. 2006. PMID: 16893904
-
Systemic delivery of P42 peptide: a new weapon to fight Huntington's disease.Acta Neuropathol Commun. 2014 Aug 5;2:86. doi: 10.1186/s40478-014-0086-x. Acta Neuropathol Commun. 2014. PMID: 25091984 Free PMC article.
-
Improvement of BDNF signalling by P42 peptide in Huntington's disease.Hum Mol Genet. 2018 Sep 1;27(17):3012-3028. doi: 10.1093/hmg/ddy207. Hum Mol Genet. 2018. PMID: 29860423
-
The emerging role of the first 17 amino acids of huntingtin in Huntington's disease.Biomol Concepts. 2015 Mar;6(1):33-46. doi: 10.1515/bmc-2015-0001. Biomol Concepts. 2015. PMID: 25741791 Free PMC article. Review.
Cited by
-
Highly Potent Peptide Therapeutics To Prevent Protein Aggregation in Huntington's Disease.ACS Med Chem Lett. 2023 Nov 14;14(12):1821-1826. doi: 10.1021/acsmedchemlett.3c00415. eCollection 2023 Dec 14. ACS Med Chem Lett. 2023. PMID: 38116434 Free PMC article.
-
Therapeutic Advances for Huntington's Disease.Brain Sci. 2020 Jan 12;10(1):43. doi: 10.3390/brainsci10010043. Brain Sci. 2020. PMID: 31940909 Free PMC article. Review.
-
Discovery of a potent small molecule inhibiting Huntington's disease (HD) pathogenesis via targeting CAG repeats RNA and Poly Q protein.Sci Rep. 2019 Nov 14;9(1):16872. doi: 10.1038/s41598-019-53410-z. Sci Rep. 2019. PMID: 31728006 Free PMC article.
-
ASPsiRNA: A Resource of ASP-siRNAs Having Therapeutic Potential for Human Genetic Disorders and Algorithm for Prediction of Their Inhibitory Efficacy.G3 (Bethesda). 2017 Sep 7;7(9):2931-2943. doi: 10.1534/g3.117.044024. G3 (Bethesda). 2017. PMID: 28696921 Free PMC article.
-
In silico designing of putative peptides for targeting pathological protein Htt in Huntington's disease.Heliyon. 2021 Feb 12;7(2):e06088. doi: 10.1016/j.heliyon.2021.e06088. eCollection 2021 Feb. Heliyon. 2021. PMID: 33659724 Free PMC article. Review.
References
-
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72:971–983 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
