

Dissecting How CD4 T Cells Are Lost During HIV Infection
- PMID: 26962940
- PMCID: PMC4835240
- DOI: 10.1016/j.chom.2016.02.012
Dissecting How CD4 T Cells Are Lost During HIV Infection
Abstract
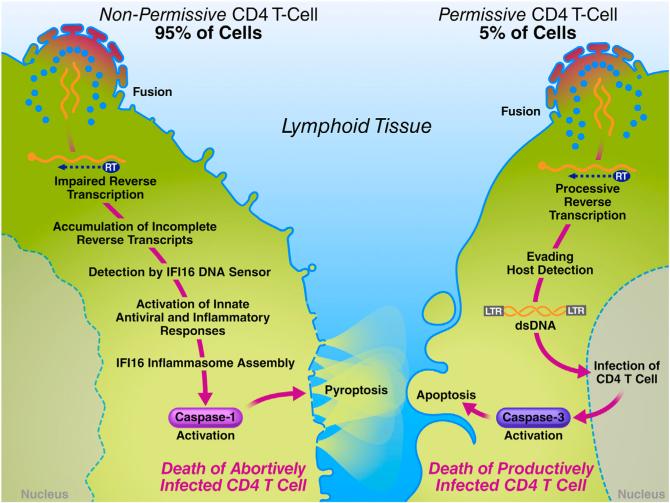
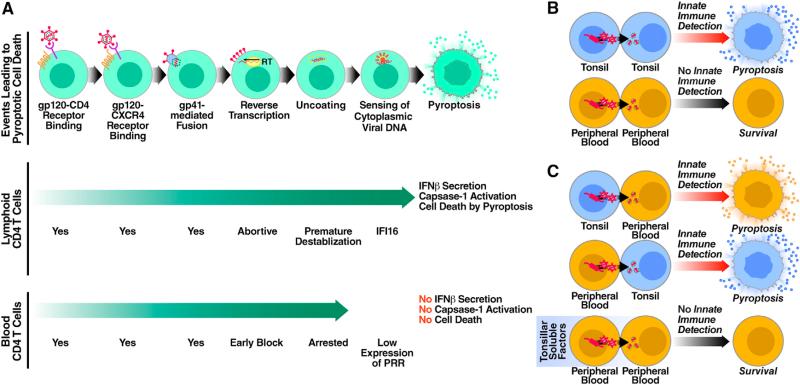
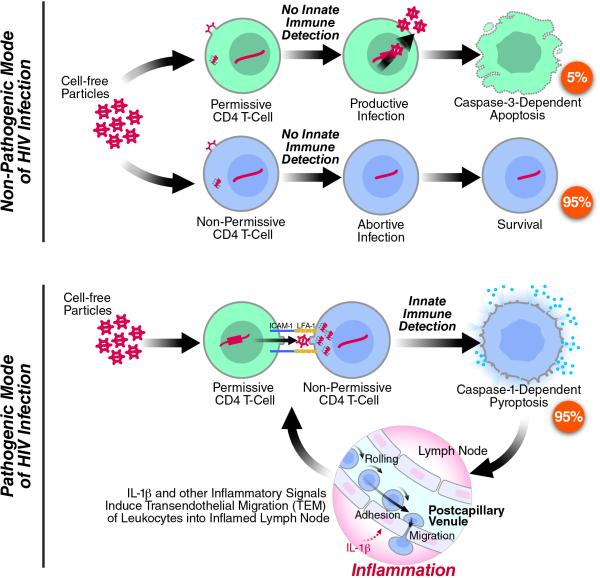
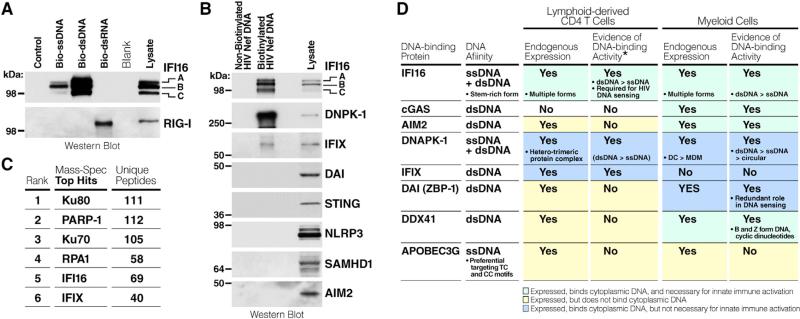
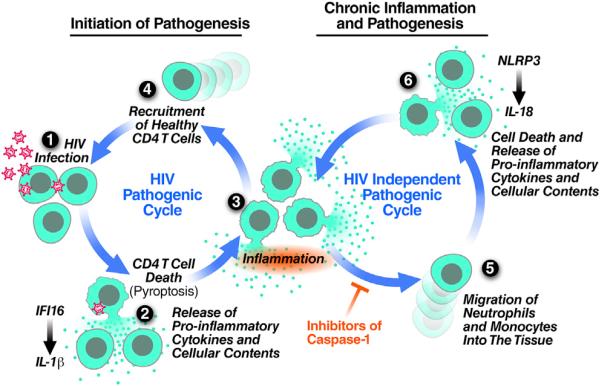
Although the replicative life cycle of HIV within CD4 T cells is understood in molecular detail, less is known about how this human retrovirus promotes the loss of CD4 T lymphocytes. It is this cell death process that drives clinical progression to acquired immune deficiency syndrome (AIDS). Recent studies have highlighted how abortive infection of resting and thus nonpermissive CD4 T cells in lymphoid tissues triggers a lethal innate immune response against the incomplete DNA products generated by inefficient viral reverse transcription in these cells. Sensing of these DNA fragments results in pyroptosis, a highly inflammatory form of programmed cell death, that potentially further perpetuates chronic inflammation and immune activation. As discussed here, these studies cast CD4 T cell death during HIV infection in a different light. Further, they identify drug targets that may be exploited to both block CD4 T cell demise and the chronic inflammatory response generated during pyroptosis.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
HIV-2 Depletes CD4 T Cells through Pyroptosis despite Vpx-Dependent Degradation of SAMHD1.J Virol. 2019 Nov 26;93(24):e00666-19. doi: 10.1128/JVI.00666-19. Print 2019 Dec 15. J Virol. 2019. PMID: 31578293 Free PMC article.
-
Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells.Cell Rep. 2015 Sep 8;12(10):1555-1563. doi: 10.1016/j.celrep.2015.08.011. Epub 2015 Aug 28. Cell Rep. 2015. PMID: 26321639 Free PMC article.
-
Next-Generation mRNA Sequencing Reveals Pyroptosis-Induced CD4+ T Cell Death in Early Simian Immunodeficiency Virus-Infected Lymphoid Tissues.J Virol. 2015 Nov 11;90(2):1080-7. doi: 10.1128/JVI.02297-15. Print 2016 Jan 15. J Virol. 2015. PMID: 26559826 Free PMC article.
-
The role of pyroptosis in incomplete immune reconstitution among people living with HIV:Potential therapeutic targets.Pharmacol Res. 2023 Nov;197:106969. doi: 10.1016/j.phrs.2023.106969. Epub 2023 Oct 21. Pharmacol Res. 2023. PMID: 37866704 Review.
-
Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues.Annu Rev Immunol. 1999;17:625-56. doi: 10.1146/annurev.immunol.17.1.625. Annu Rev Immunol. 1999. PMID: 10358770 Review.
Cited by
-
Simian Immunodeficiency Virus Infection Mediated Changes in Jejunum and Peripheral SARS-CoV-2 Receptor ACE2 and Associated Proteins or Genes in Rhesus Macaques.Front Immunol. 2022 Feb 25;13:835686. doi: 10.3389/fimmu.2022.835686. eCollection 2022. Front Immunol. 2022. PMID: 35281029 Free PMC article.
-
Cell pyroptosis in health and inflammatory diseases.Cell Death Discov. 2022 Apr 11;8(1):191. doi: 10.1038/s41420-022-00998-3. Cell Death Discov. 2022. PMID: 35411030 Free PMC article.
-
Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease.J Clin Med. 2020 May 12;9(5):1440. doi: 10.3390/jcm9051440. J Clin Med. 2020. PMID: 32408624 Free PMC article. Review.
-
The role of pyroptosis in viral infection.Arch Virol. 2024 Mar 8;169(3):69. doi: 10.1007/s00705-024-05978-9. Arch Virol. 2024. PMID: 38456965 Review.
-
Exercise to Prevent Accelerated Vascular Aging in People Living With HIV.Circ Res. 2024 May 24;134(11):1607-1635. doi: 10.1161/CIRCRESAHA.124.323975. Epub 2024 May 23. Circ Res. 2024. PMID: 38781293 Review.
References
-
- Alimonti JB, Ball TB, Fowke KR. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J. Gen. Virol. 2003;84:1649–1661. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
