

SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3β
- PMID: 26667039
- PMCID: PMC4760222
- DOI: 10.1128/MCB.00586-15
SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3β
Abstract
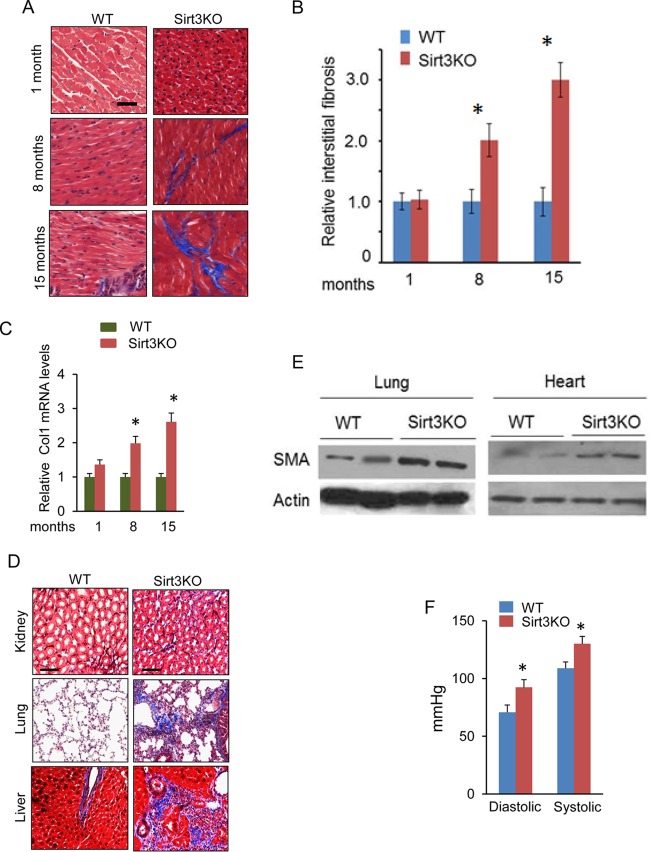
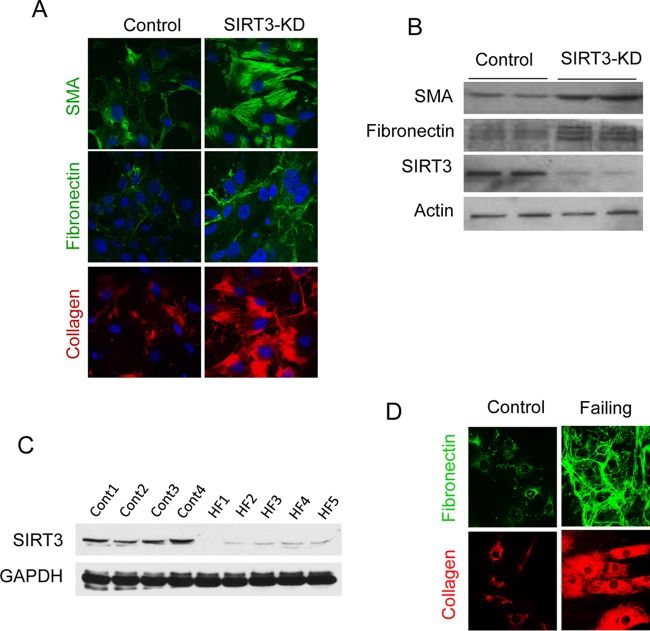
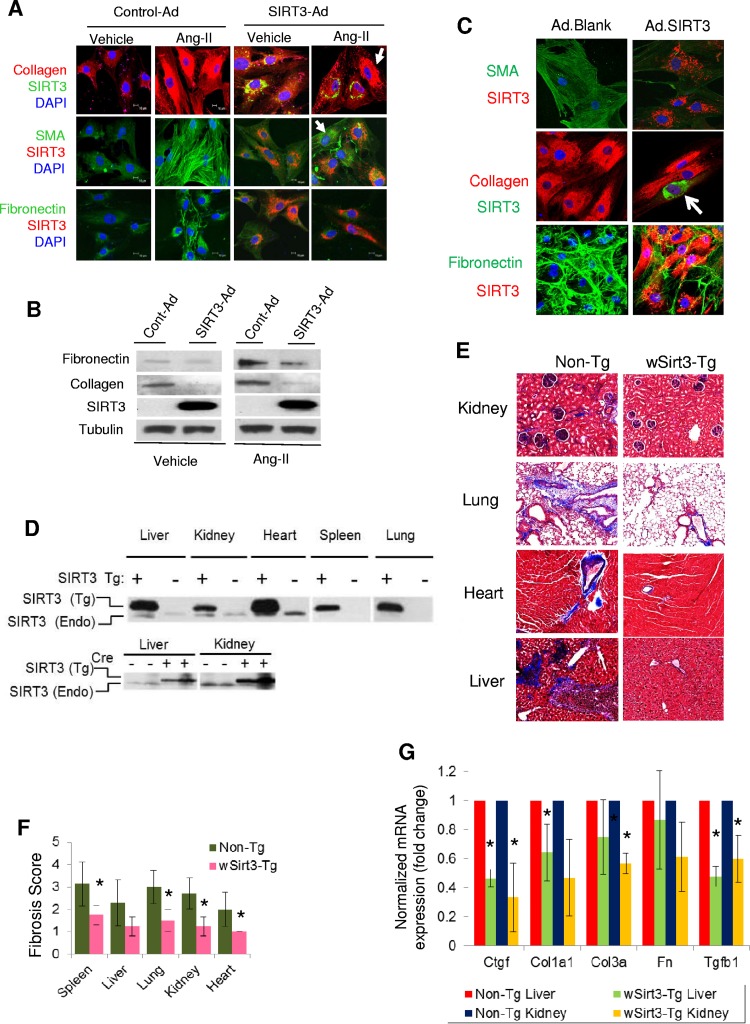
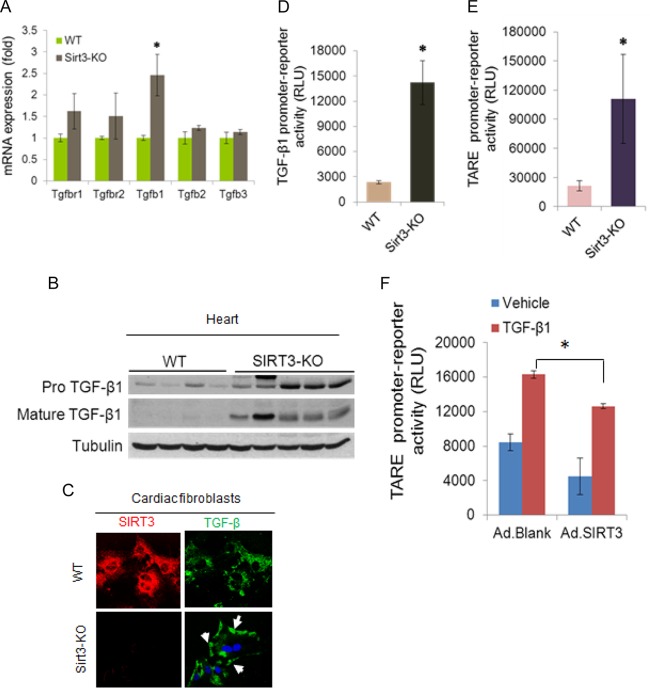
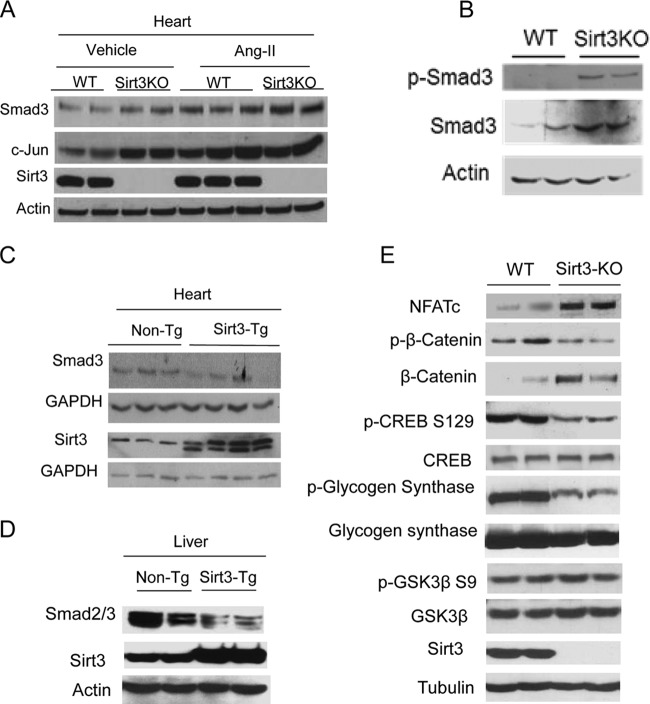
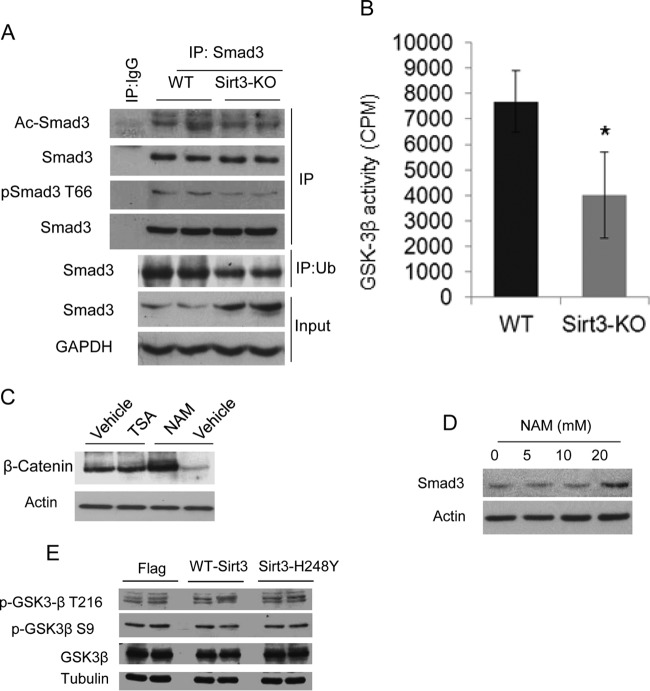
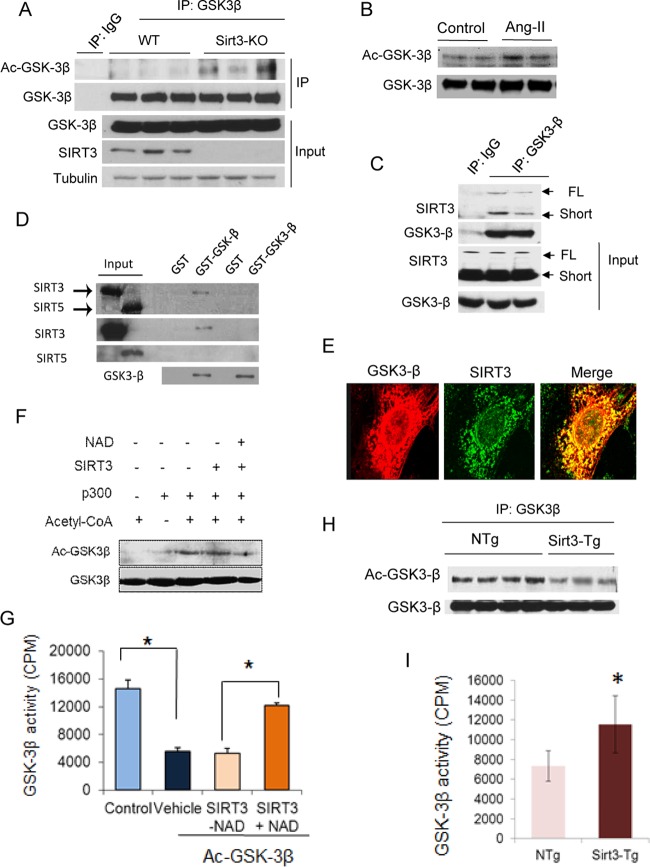
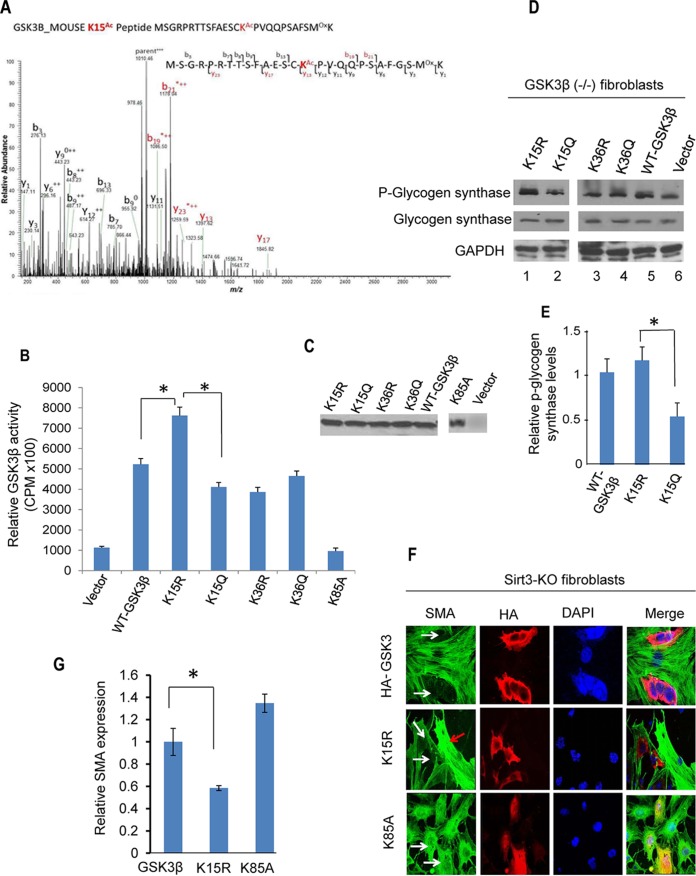
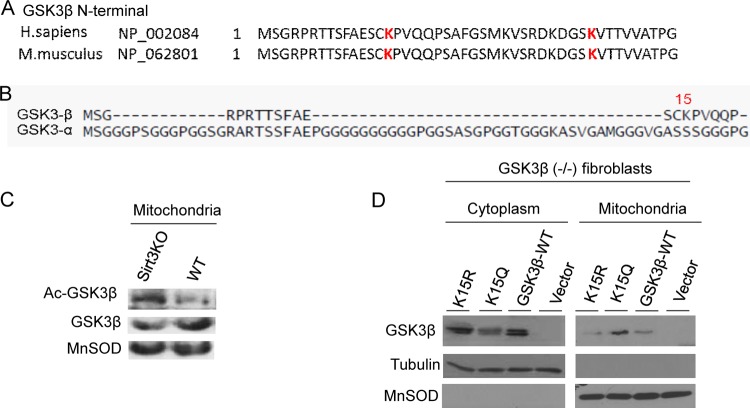
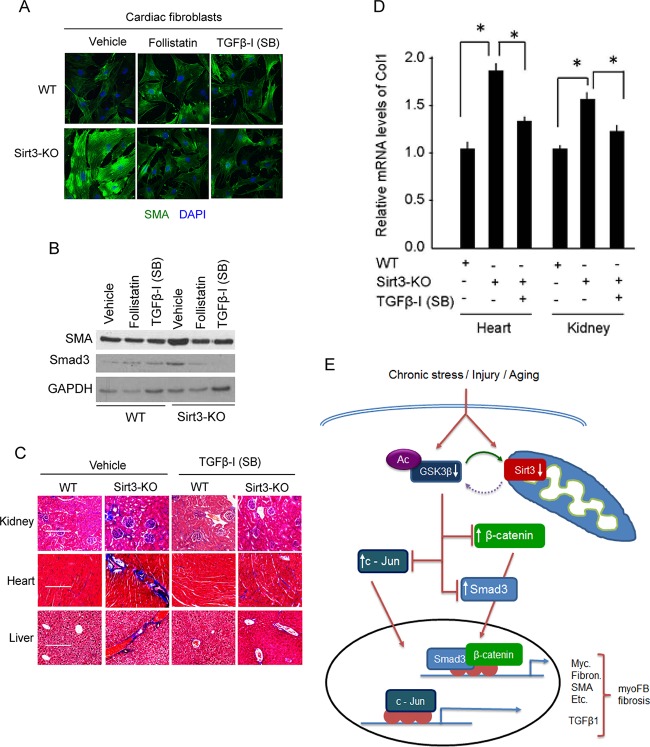
Tissue fibrosis is a major cause of organ dysfunction during chronic diseases and aging. A critical step in this process is transforming growth factor β1 (TGF-β1)-mediated transformation of fibroblasts into myofibroblasts, cells capable of synthesizing extracellular matrix. Here, we show that SIRT3 controls transformation of fibroblasts into myofibroblasts via suppressing the profibrotic TGF-β1 signaling. We found that Sirt3 knockout (KO) mice with age develop tissue fibrosis of multiple organs, including heart, liver, kidney, and lungs but not whole-body SIRT3-overexpressing mice. SIRT3 deficiency caused induction of TGF-β1 expression and hyperacetylation of glycogen synthase kinase 3β (GSK3β) at residue K15, which negatively regulated GSK3β activity to phosphorylate the substrates Smad3 and β-catenin. Reduced phosphorylation led to stabilization and activation of these transcription factors regulating expression of the profibrotic genes. SIRT3 deacetylated and activated GSK3β and thereby blocked TGF-β1 signaling and tissue fibrosis. These data reveal a new role of SIRT3 to negatively regulate aging-associated tissue fibrosis and discloses a novel phosphorylation-independent mechanism controlling the catalytic activity of GSK3β.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia-reperfusion in mice.Dis Model Mech. 2015 Aug 1;8(8):931-40. doi: 10.1242/dmm.020511. Epub 2015 Jun 18. Dis Model Mech. 2015. PMID: 26092126 Free PMC article.
-
Sirtuin 6 deficiency transcriptionally up-regulates TGF-β signaling and induces fibrosis in mice.J Biol Chem. 2020 Jan 10;295(2):415-434. doi: 10.1074/jbc.RA118.007212. Epub 2019 Nov 19. J Biol Chem. 2020. PMID: 31744885 Free PMC article.
-
TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts.Pharmacol Res. 2008 Apr;57(4):274-82. doi: 10.1016/j.phrs.2008.02.001. Epub 2008 Feb 9. Pharmacol Res. 2008. PMID: 18346908
-
Entanglement of GSK-3β, β-catenin and TGF-β1 signaling network to regulate myocardial fibrosis.J Mol Cell Cardiol. 2017 Sep;110:109-120. doi: 10.1016/j.yjmcc.2017.07.011. Epub 2017 Jul 27. J Mol Cell Cardiol. 2017. PMID: 28756206 Free PMC article. Review.
-
Hyaluronan's Role in Fibrosis: A Pathogenic Factor or a Passive Player?Biomed Res Int. 2015;2015:790203. doi: 10.1155/2015/790203. Epub 2015 Oct 25. Biomed Res Int. 2015. PMID: 26583132 Free PMC article. Review.
Cited by
-
NBD-based synthetic probes for sensing small molecules and proteins: design, sensing mechanisms and biological applications.Chem Soc Rev. 2021 Jul 5;50(13):7436-7495. doi: 10.1039/d0cs01096k. Chem Soc Rev. 2021. PMID: 34075930 Free PMC article. Review.
-
The Role of Mitochondrial Sirtuins (SIRT3, SIRT4 and SIRT5) in Renal Cell Metabolism: Implication for Kidney Diseases.Int J Mol Sci. 2024 Jun 25;25(13):6936. doi: 10.3390/ijms25136936. Int J Mol Sci. 2024. PMID: 39000044 Free PMC article. Review.
-
SIRT3/6: an amazing challenge and opportunity in the fight against fibrosis and aging.Cell Mol Life Sci. 2024 Jan 31;81(1):69. doi: 10.1007/s00018-023-05093-z. Cell Mol Life Sci. 2024. PMID: 38294557 Free PMC article. Review.
-
Aging, longevity, and the role of environmental stressors: a focus on wildfire smoke and air quality.Front Toxicol. 2023 Oct 11;5:1267667. doi: 10.3389/ftox.2023.1267667. eCollection 2023. Front Toxicol. 2023. PMID: 37900096 Free PMC article. Review.
-
Mitochondrial Quality Control in Cardiomyocytes: A Critical Role in the Progression of Cardiovascular Diseases.Front Physiol. 2020 Mar 27;11:252. doi: 10.3389/fphys.2020.00252. eCollection 2020. Front Physiol. 2020. PMID: 32292354 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous