

Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene
- PMID: 26482871
- PMCID: PMC4695917
- DOI: 10.1016/j.clim.2015.10.002
Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene
Abstract
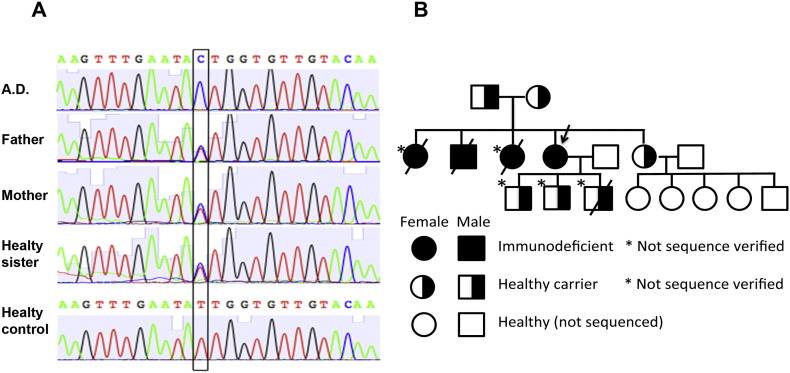
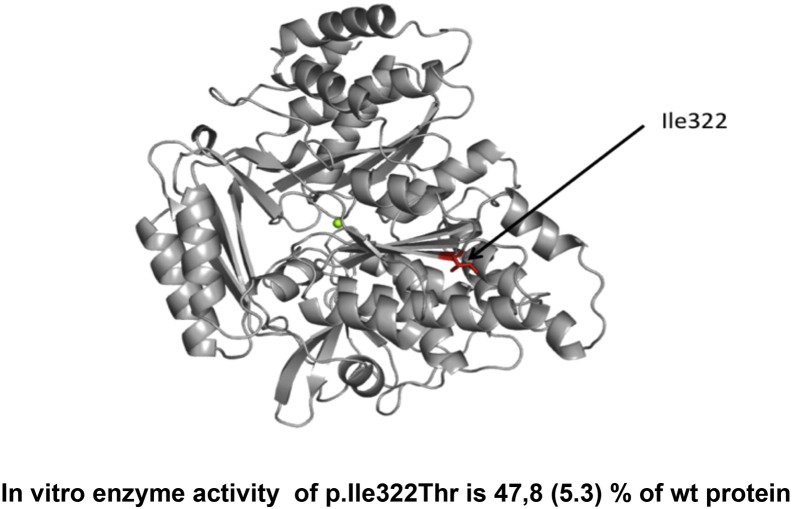
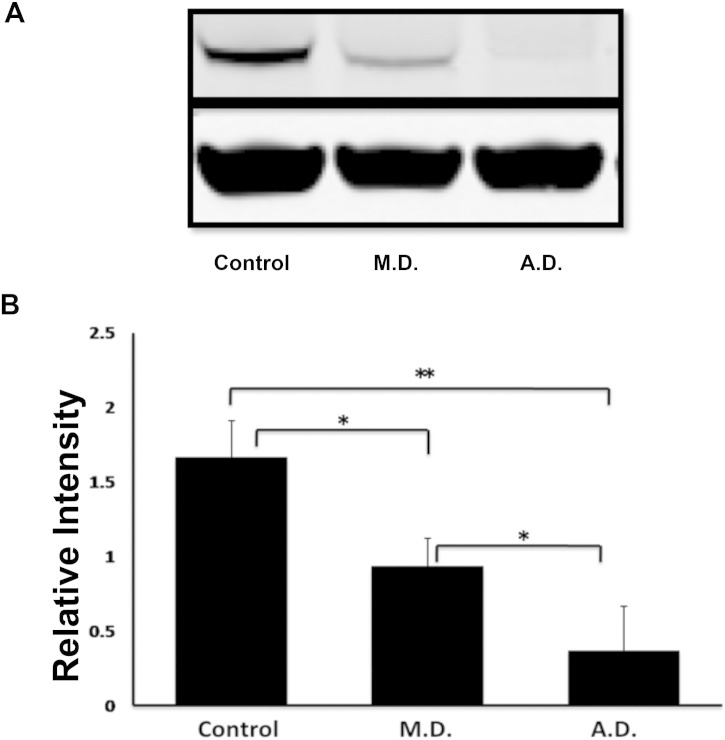
Phosphoglucomutase 3 (PGM3) is an enzyme converting N-acetyl-glucosamine-6-phosphate to N-acetyl-glucosamine-1-phosphate, a precursor important for glycosylation. Mutations in the PGM3 gene have recently been identified as the cause of novel primary immunodeficiency with a hyper-IgE like syndrome. Here we report the occurrence of a homozygous mutation in the PGM3 gene in a family with immunodeficient children, described already in 1976. DNA from two of the immunodeficient siblings was sequenced and shown to encode the same homozygous missense mutation, causing a destabilized protein with reduced enzymatic capacity. Affected individuals were highly prone to infections, but lack the developmental defects in the nervous and skeletal systems, reported in other families. Moreover, normal IgE levels were found. Thus, belonging to the expanding group of congenital glycosylation defects, PGM3 deficiency is characterized by immunodeficiency, with or without increased IgE levels, and with variable forms of developmental defects affecting other organ systems.
Keywords: CDG; Congenital defects of glycosylation; N-acetylglucosamine-phosphate mutase; Primary immunodeficiency; hyper-IgE syndrome.
Figures
Similar articles
-
Eleven percent intact PGM3 in a severely immunodeficient patient with a novel splice-site mutation, a case report.BMC Pediatr. 2018 Aug 29;18(1):285. doi: 10.1186/s12887-018-1258-9. BMC Pediatr. 2018. PMID: 30157810 Free PMC article.
-
Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels.J Allergy Clin Immunol. 2014 May;133(5):1410-9, 1419.e1-13. doi: 10.1016/j.jaci.2014.02.025. Epub 2014 Apr 1. J Allergy Clin Immunol. 2014. PMID: 24698316 Free PMC article. Clinical Trial.
-
Hyper-IgE syndromes: reviewing PGM3 deficiency.Curr Opin Pediatr. 2014 Dec;26(6):697-703. doi: 10.1097/MOP.0000000000000158. Curr Opin Pediatr. 2014. PMID: 25365149 Review.
-
A founder mutation underlies a severe form of phosphoglutamase 3 (PGM3) deficiency in Tunisian patients.Mol Immunol. 2017 Oct;90:57-63. doi: 10.1016/j.molimm.2017.06.248. Epub 2017 Jul 10. Mol Immunol. 2017. PMID: 28704707
-
Hyper-IgE Syndromes and the Lung.Clin Chest Med. 2016 Sep;37(3):557-67. doi: 10.1016/j.ccm.2016.04.016. Epub 2016 Jun 10. Clin Chest Med. 2016. PMID: 27514600 Free PMC article. Review.
Cited by
-
A De Novo Cause of PGM3 Deficiency Treated with Hematopoietic Stem Cell Transplantation.J Clin Immunol. 2022 Apr;42(3):691-694. doi: 10.1007/s10875-021-01196-z. Epub 2022 Jan 18. J Clin Immunol. 2022. PMID: 35040011 Free PMC article. No abstract available.
-
Heterozygous PGM3 Variants Are Associated With Idiopathic Focal Epilepsy With Incomplete Penetrance.Front Genet. 2020 Oct 15;11:559080. doi: 10.3389/fgene.2020.559080. eCollection 2020. Front Genet. 2020. PMID: 33193641 Free PMC article.
-
CDG Therapies: From Bench to Bedside.Int J Mol Sci. 2018 Apr 27;19(5):1304. doi: 10.3390/ijms19051304. Int J Mol Sci. 2018. PMID: 29702557 Free PMC article. Review.
-
Clinical Utility Gene Card for: PGM3 defective congenital disorder of glycosylation.Eur J Hum Genet. 2019 Nov;27(11):1757-1760. doi: 10.1038/s41431-019-0453-y. Epub 2019 Jun 23. Eur J Hum Genet. 2019. PMID: 31231132 Free PMC article.
-
What is new in CDG?J Inherit Metab Dis. 2017 Jul;40(4):569-586. doi: 10.1007/s10545-017-0050-6. Epub 2017 May 8. J Inherit Metab Dis. 2017. PMID: 28484880 Review.
References
-
- Pang H., Koda Y., Soejima M., Kimura H. Identification of human phosphoglucomutase 3 (PGM3) as N-acetylglucosamine-phosphate mutase (AGM1) Ann. Hum. Genet. 2002;66:139–144. - PubMed
-
- Ohtsubo K., Marth J.D. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. - PubMed
-
- Kreppel L.K., Hart G.W. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J. Biol. Chem. 1999;274:32015–32022. - PubMed
-
- Wells L., Vosseller K., Hart G.W. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291:2376–2378. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
