

Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation
- PMID: 26302407
- PMCID: PMC4699446
- DOI: 10.1038/ncb3228
Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation
Erratum in
-
Corrigendum: Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation.Nat Cell Biol. 2015 Oct;17(10):1370. doi: 10.1038/ncb3242. Nat Cell Biol. 2015. PMID: 26419803 No abstract available.
Abstract
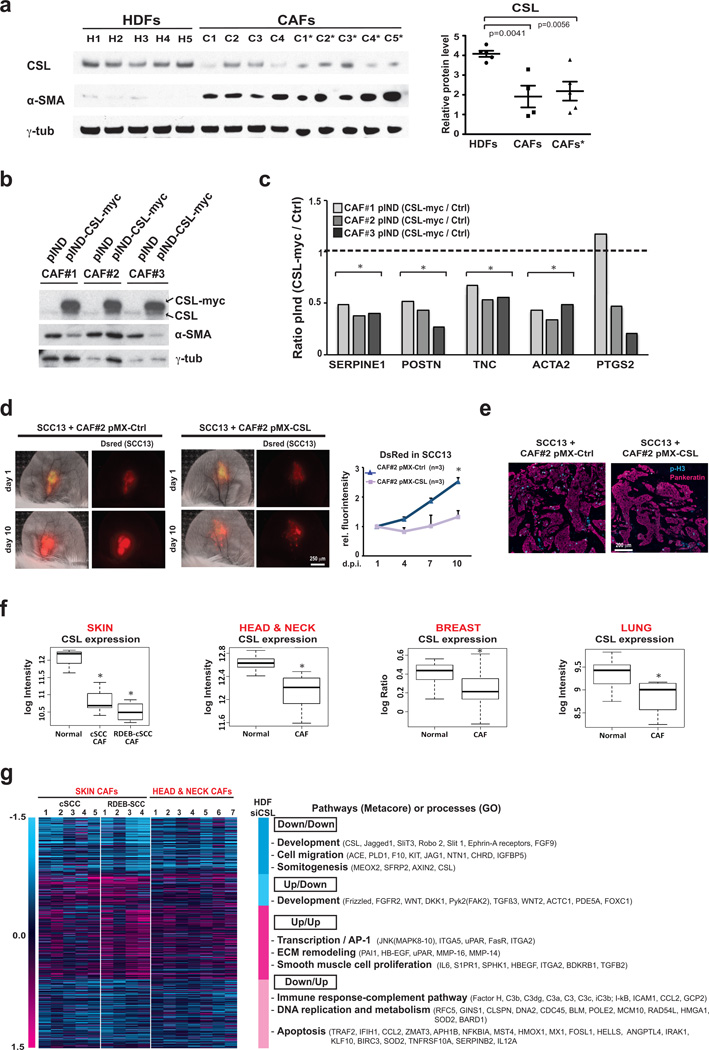
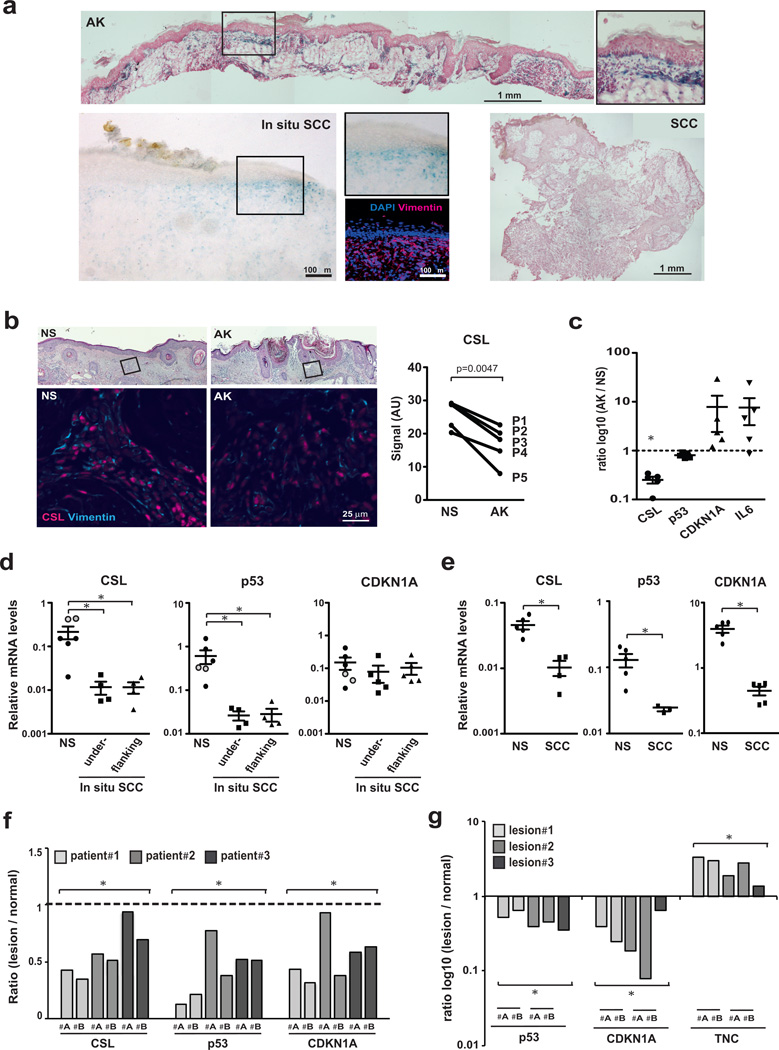
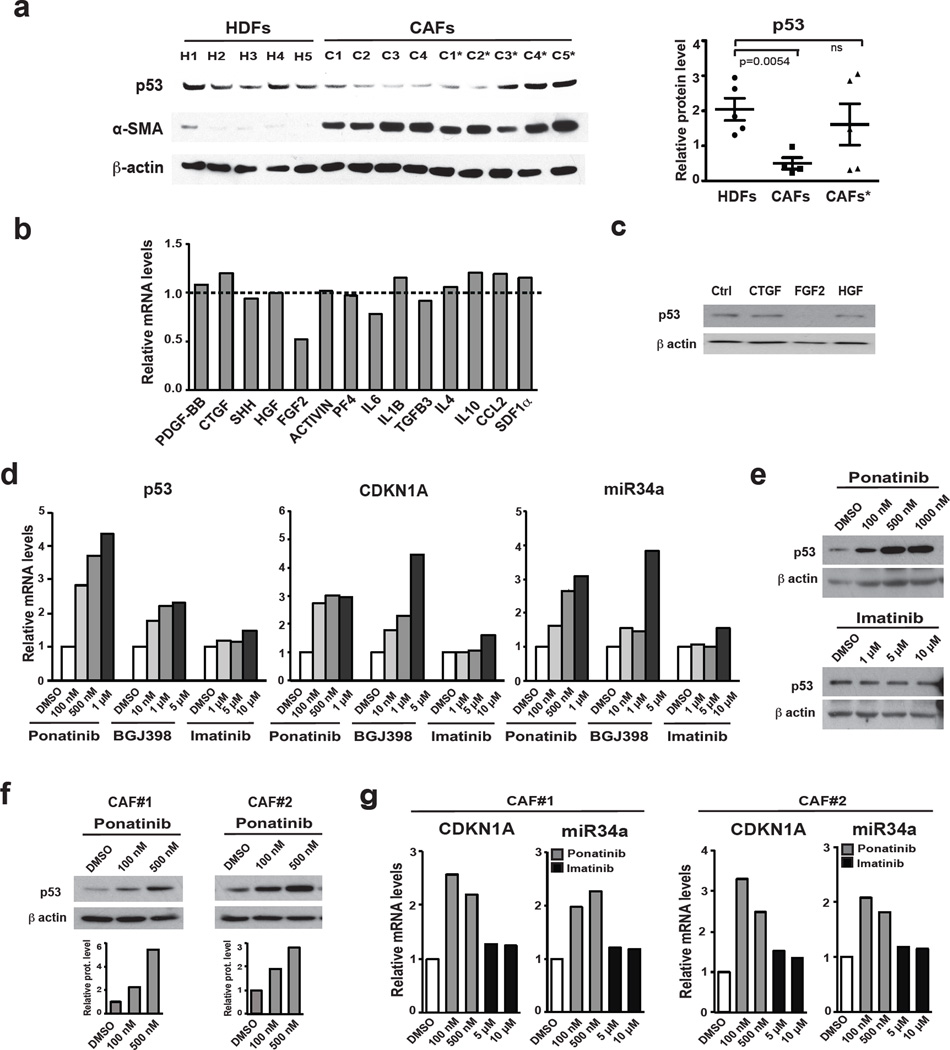
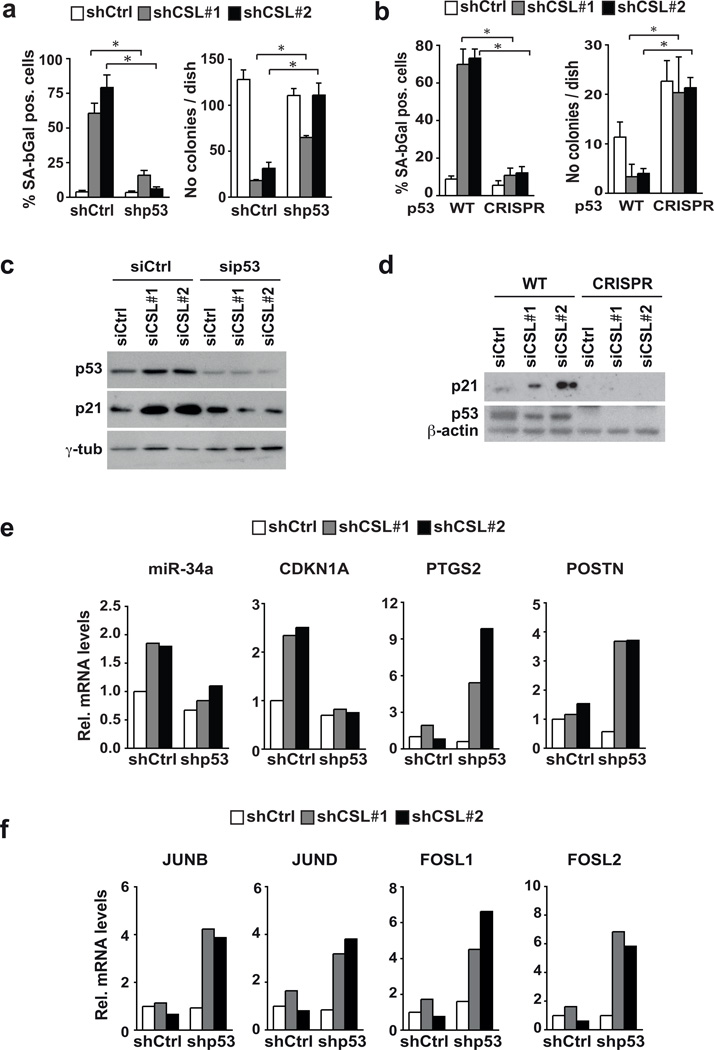
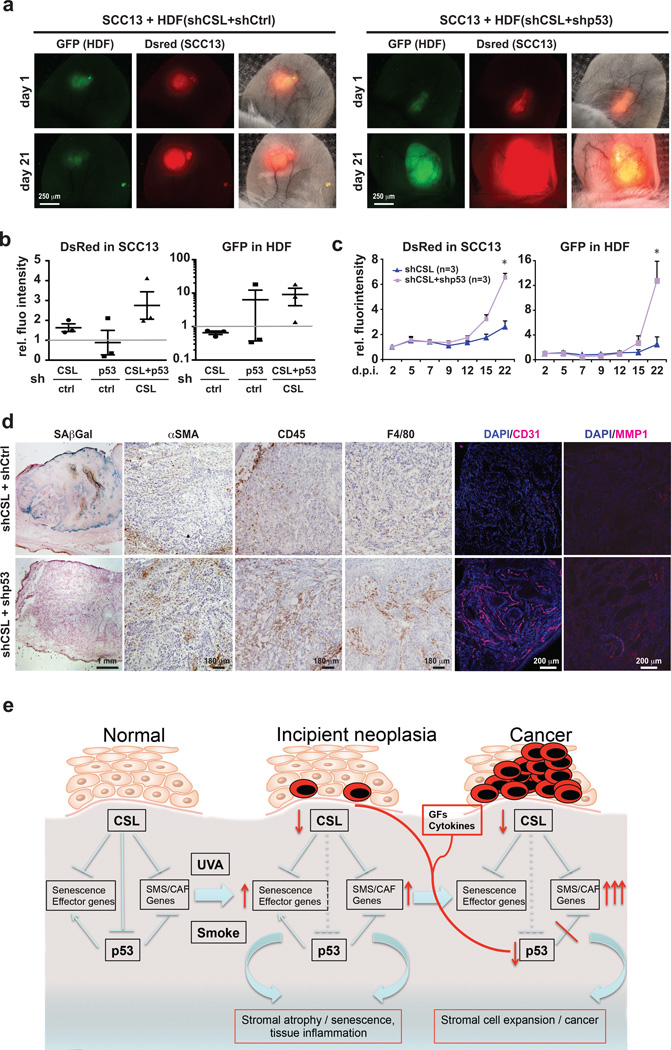
Stromal fibroblast senescence has been linked to ageing-associated cancer risk. However, density and proliferation of cancer-associated fibroblasts (CAFs) are frequently increased. Loss or downmodulation of the Notch effector CSL (also known as RBP-Jκ) in dermal fibroblasts is sufficient for CAF activation and ensuing keratinocyte-derived tumours. We report that CSL silencing induces senescence of primary fibroblasts from dermis, oral mucosa, breast and lung. CSL functions in these cells as a direct repressor of multiple senescence- and CAF-effector genes. It also physically interacts with p53, repressing its activity. CSL is downmodulated in stromal fibroblasts of premalignant skin actinic keratosis lesions and squamous cell carcinomas, whereas p53 expression and function are downmodulated only in the latter, with paracrine FGF signalling as the probable culprit. Concomitant loss of CSL and p53 overcomes fibroblast senescence, enhances expression of CAF effectors and promotes stromal and cancer cell expansion. The findings support a CAF activation-stromal co-evolution model under convergent CSL-p53 control.
Figures



Similar articles
-
Androgen receptor functions as transcriptional repressor of cancer-associated fibroblast activation.J Clin Invest. 2018 Dec 3;128(12):5531-5548. doi: 10.1172/JCI99159. Epub 2018 Nov 5. J Clin Invest. 2018. PMID: 30395538 Free PMC article.
-
PDCD4 is a CSL associated protein with a transcription repressive function in cancer associated fibroblast activation.Oncotarget. 2016 Sep 13;7(37):58717-58727. doi: 10.18632/oncotarget.11227. Oncotarget. 2016. PMID: 27542230 Free PMC article.
-
Notch-effector CSL promotes squamous cell carcinoma by repressing histone demethylase KDM6B.J Clin Invest. 2018 Jun 1;128(6):2581-2599. doi: 10.1172/JCI96915. Epub 2018 May 14. J Clin Invest. 2018. PMID: 29757189 Free PMC article.
-
Role of apoptosis in basal cell and squamous cell carcinoma formation.Immunol Lett. 2005 Aug 15;100(1):68-72. doi: 10.1016/j.imlet.2005.06.008. Immunol Lett. 2005. PMID: 16054233 Review.
-
The effects of conformational heterogeneity on the binding of the Notch intracellular domain to effector proteins: a case of biologically tuned disorder.Biochem Soc Trans. 2008 Apr;36(Pt 2):157-66. doi: 10.1042/BST0360157. Biochem Soc Trans. 2008. PMID: 18363556 Review.
Cited by
-
Can Targeting Stroma Pave the Way to Enhanced Antitumor Immunity and Immunotherapy of Solid Tumors?Cancer Immunol Res. 2016 Apr;4(4):269-78. doi: 10.1158/2326-6066.CIR-16-0011. Cancer Immunol Res. 2016. PMID: 27036971 Free PMC article. Review.
-
Cold Atmospheric Plasma (CAP) for the Treatment of Actinic Keratosis and Skin Field Cancerization: Clinical and High-Frequency Ultrasound Evaluation.Dermatol Ther (Heidelb). 2021 Jun;11(3):855-866. doi: 10.1007/s13555-021-00514-y. Epub 2021 Mar 18. Dermatol Ther (Heidelb). 2021. PMID: 33738749 Free PMC article.
-
Microenvironmental modulation of the developing tumour: an immune-stromal dialogue.Mol Oncol. 2021 Oct;15(10):2600-2633. doi: 10.1002/1878-0261.12773. Epub 2020 Aug 28. Mol Oncol. 2021. PMID: 32741067 Free PMC article. Review.
-
Effect of Anlotinib as a Third-Line or Further Treatment on Overall Survival of Patients With Advanced Non-Small Cell Lung Cancer: The ALTER 0303 Phase 3 Randomized Clinical Trial.JAMA Oncol. 2018 Nov 1;4(11):1569-1575. doi: 10.1001/jamaoncol.2018.3039. JAMA Oncol. 2018. PMID: 30098152 Free PMC article. Clinical Trial.
-
Androgen receptor functions as transcriptional repressor of cancer-associated fibroblast activation.J Clin Invest. 2018 Dec 3;128(12):5531-5548. doi: 10.1172/JCI99159. Epub 2018 Nov 5. J Clin Invest. 2018. PMID: 30395538 Free PMC article.
References
-
- Gatenby R. Perspective: Finding cancer's first principles. Nature. 2012;491:S55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
