

Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy
- PMID: 25892230
- PMCID: PMC4438771
- DOI: 10.1016/j.celrep.2015.03.055
Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy
Abstract
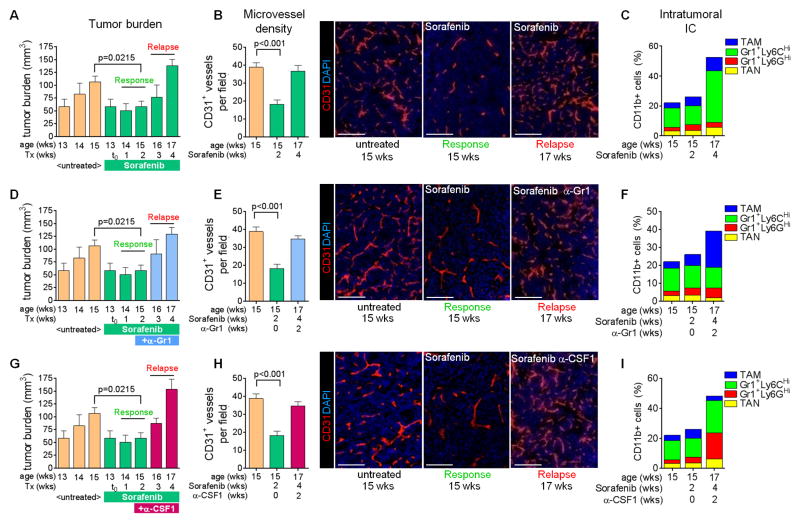
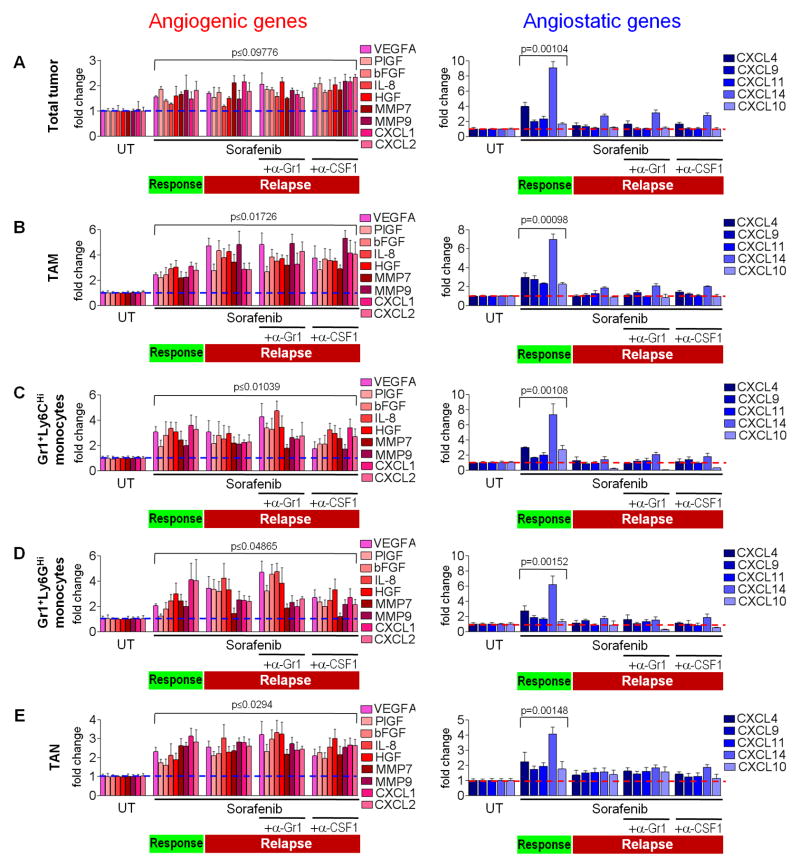
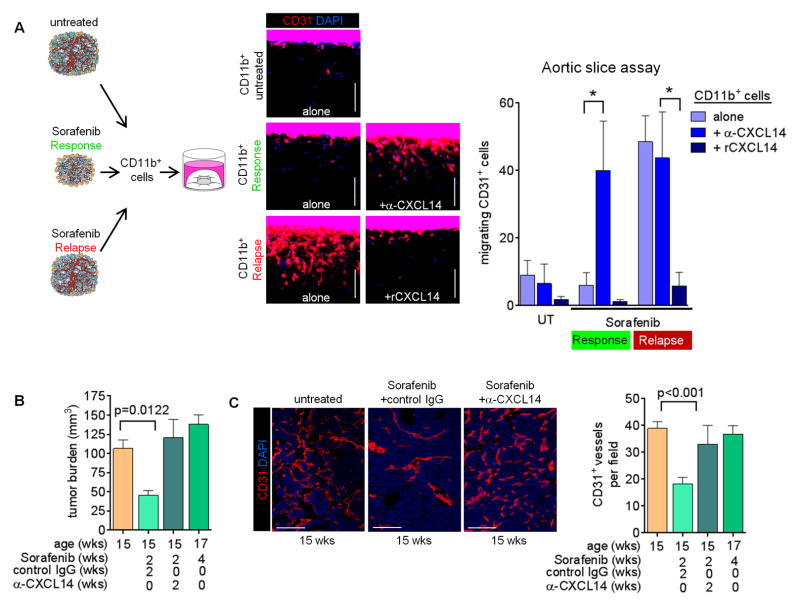
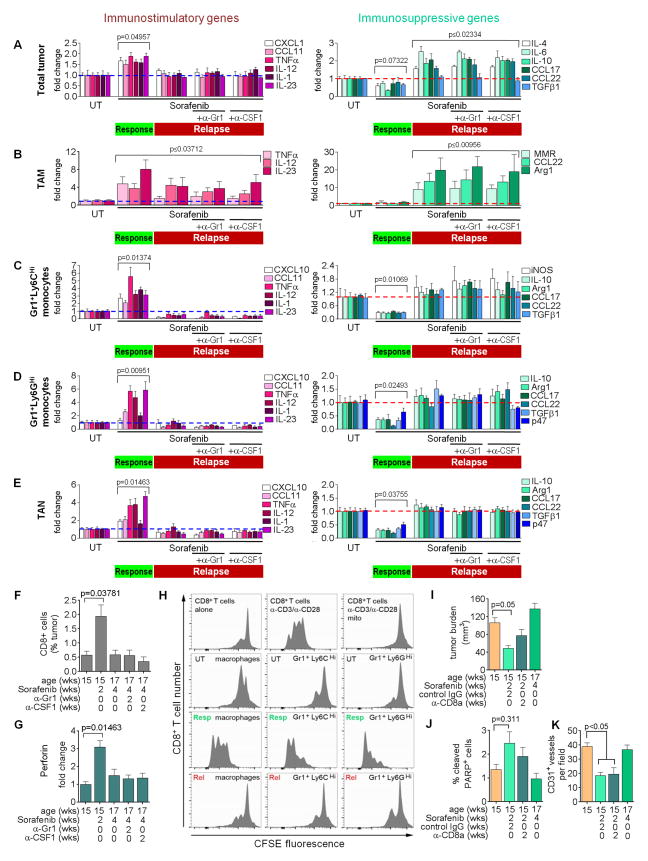
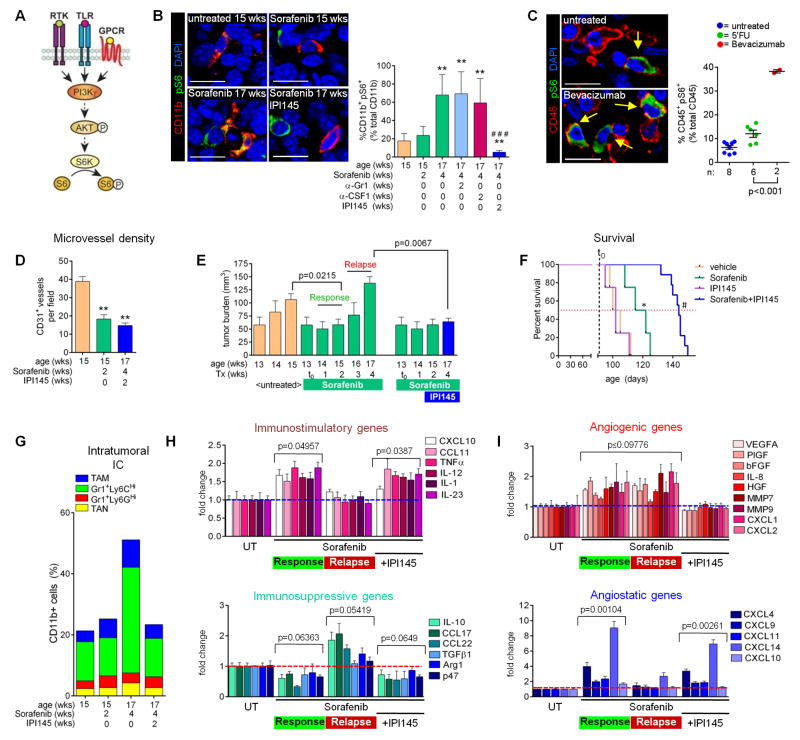
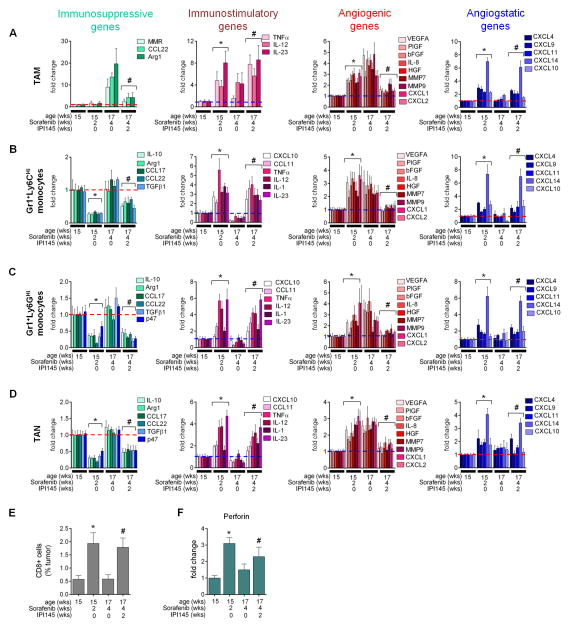
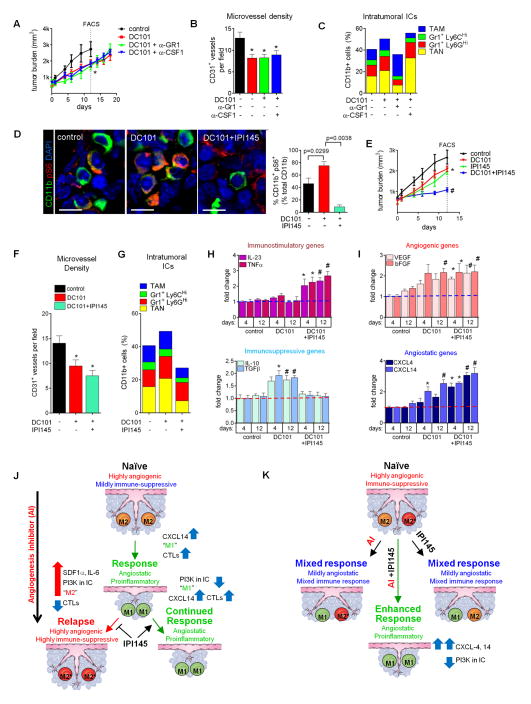
Antiangiogenic therapy is commonly used in the clinic, but its beneficial effects are short-lived, leading to tumor relapse within months. Here, we found that the efficacy of angiogenic inhibitors targeting the VEGF/VEGFR pathway was dependent on induction of the angiostatic and immune-stimulatory chemokine CXCL14 in mouse models of pancreatic neuroendocrine and mammary tumors. In response, tumors reinitiated angiogenesis and immune suppression by activating PI3K signaling in all CD11b+ cells, rendering tumors nonresponsive to VEGF/VEGFR inhibition. Adaptive resistance was also associated with an increase in Gr1+CD11b+ cells, but targeting Gr1+ cells was not sufficient to further sensitize angiogenic blockade because tumor-associated macrophages (TAMs) would compensate for the lack of such cells and vice versa, leading to an oscillating pattern of distinct immune-cell populations. However, PI3K inhibition in CD11b+ myeloid cells generated an enduring angiostatic and immune-stimulatory environment in which antiangiogenic therapy remained efficient.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells.Cancer Res. 2008 Jul 15;68(14):5501-4. doi: 10.1158/0008-5472.CAN-08-0925. Cancer Res. 2008. PMID: 18632597 Review.
-
mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor.Clin Cancer Res. 2009 Mar 1;15(5):1612-22. doi: 10.1158/1078-0432.CCR-08-2057. Epub 2009 Feb 17. Clin Cancer Res. 2009. PMID: 19223496
-
Imaging tumor angiogenesis.J Clin Invest. 2006 Oct;116(10):2585-7. doi: 10.1172/JCI30058. J Clin Invest. 2006. PMID: 17016553 Free PMC article.
-
Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells.Nat Biotechnol. 2007 Aug;25(8):911-20. doi: 10.1038/nbt1323. Epub 2007 Jul 29. Nat Biotechnol. 2007. PMID: 17664940
-
The Sabotaging Role of Myeloid Cells in Anti-Angiogenic Therapy: Coordination of Angiogenesis and Immune Suppression by Hypoxia.J Cell Physiol. 2017 Sep;232(9):2312-2322. doi: 10.1002/jcp.25726. Epub 2017 Apr 10. J Cell Physiol. 2017. PMID: 27935039 Review.
Cited by
-
FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review).Int J Mol Med. 2016 Jul;38(1):3-15. doi: 10.3892/ijmm.2016.2620. Epub 2016 May 31. Int J Mol Med. 2016. PMID: 27245147 Free PMC article. Review.
-
The Role of Chemokines in Orchestrating the Immune Response to Pancreatic Ductal Adenocarcinoma.Cancers (Basel). 2024 Jan 28;16(3):559. doi: 10.3390/cancers16030559. Cancers (Basel). 2024. PMID: 38339310 Free PMC article. Review.
-
Autophagy, cancer and angiogenesis: where is the link?Cell Biosci. 2019 Aug 13;9:65. doi: 10.1186/s13578-019-0327-6. eCollection 2019. Cell Biosci. 2019. PMID: 31428311 Free PMC article. Review.
-
Lenvatinib for effectively treating antiangiogenic drug-resistant nasopharyngeal carcinoma.Cell Death Dis. 2022 Aug 19;13(8):724. doi: 10.1038/s41419-022-05171-3. Cell Death Dis. 2022. PMID: 35985991 Free PMC article.
-
The reciprocal function and regulation of tumor vessels and immune cells offers new therapeutic opportunities in cancer.Semin Cancer Biol. 2018 Oct;52(Pt 2):107-116. doi: 10.1016/j.semcancer.2018.06.002. Epub 2018 Jun 20. Semin Cancer Biol. 2018. PMID: 29935312 Free PMC article. Review.
References
-
- Anisimov A, Tvorogov D, Alitalo A, Leppanen VM, An Y, Han EC, Orsenigo F, Gaal EI, Holopainen T, Koh YJ, et al. Vascular endothelial growth factor-angiopoietin chimera with improved properties for therapeutic angiogenesis. Circulation. 2013;127:424–434. - PubMed
-
- Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Seminars in cancer biology. 2009;19:329–337. - PubMed
-
- Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
