

HIV-1 integration landscape during latent and active infection
- PMID: 25635456
- PMCID: PMC4371550
- DOI: 10.1016/j.cell.2015.01.020
HIV-1 integration landscape during latent and active infection
Abstract
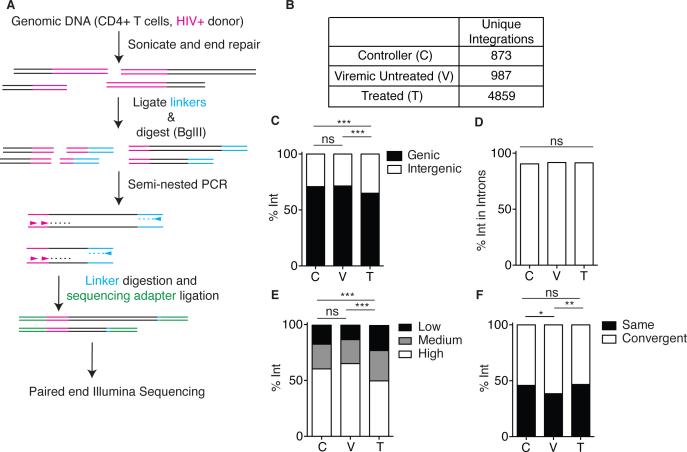
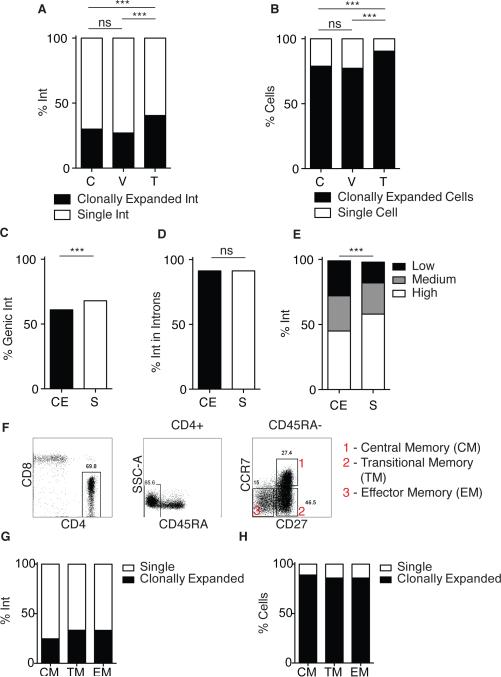
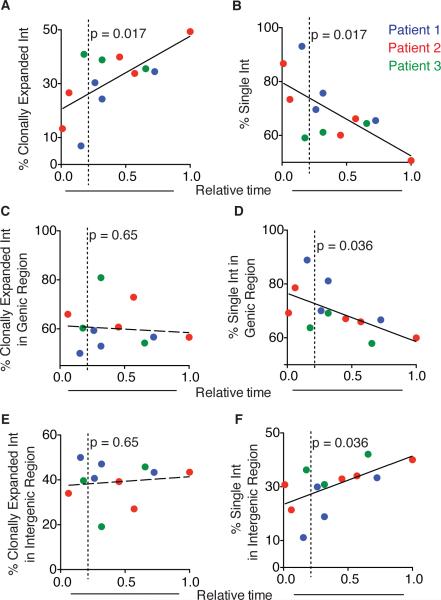
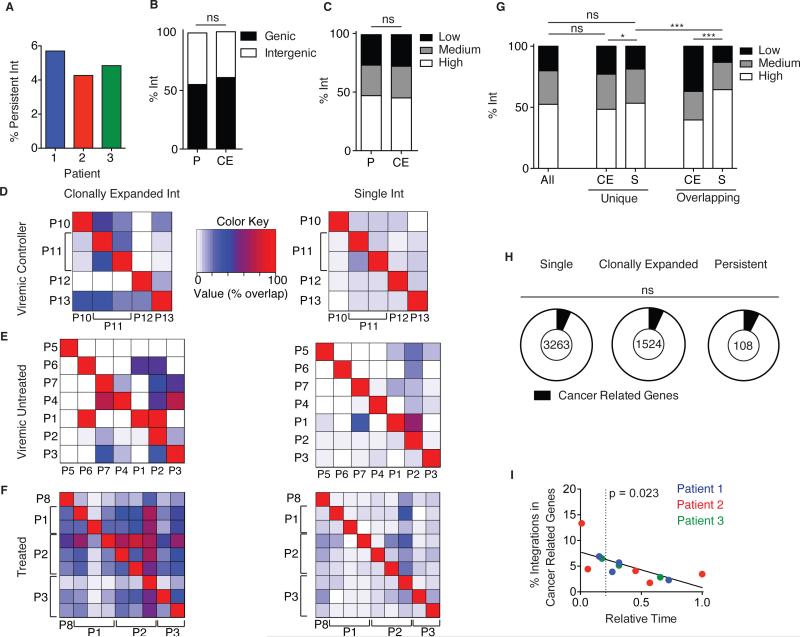
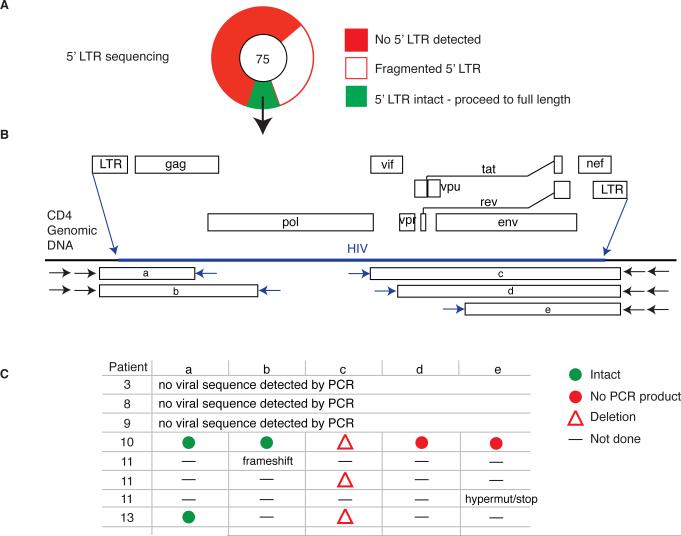
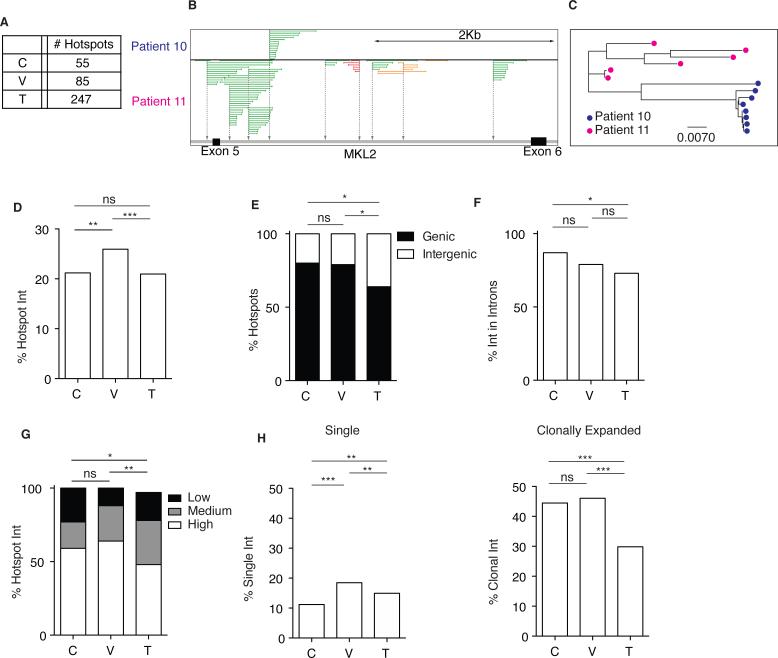
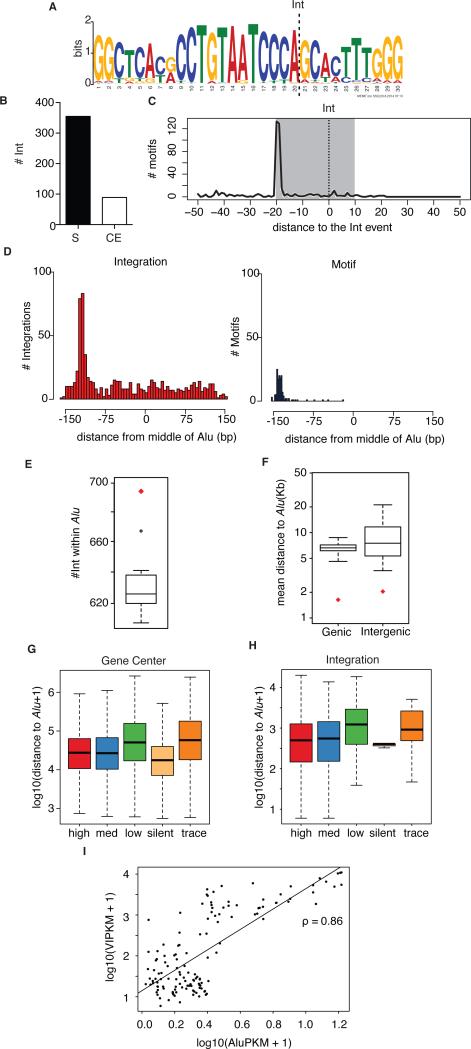
The barrier to curing HIV-1 is thought to reside primarily in CD4(+) T cells containing silent proviruses. To characterize these latently infected cells, we studied the integration profile of HIV-1 in viremic progressors, individuals receiving antiretroviral therapy, and viremic controllers. Clonally expanded T cells represented the majority of all integrations and increased during therapy. However, none of the 75 expanded T cell clones assayed contained intact virus. In contrast, the cells bearing single integration events decreased in frequency over time on therapy, and the surviving cells were enriched for HIV-1 integration in silent regions of the genome. Finally, there was a strong preference for integration into, or in close proximity to, Alu repeats, which were also enriched in local hotspots for integration. The data indicate that dividing clonally expanded T cells contain defective proviruses and that the replication-competent reservoir is primarily found in CD4(+) T cells that remain relatively quiescent.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Experimental approaches to the study of HIV-1 latency.Nat Rev Microbiol. 2007 Feb;5(2):95-106. doi: 10.1038/nrmicro1580. Nat Rev Microbiol. 2007. PMID: 17224919 Review.
-
New Approaches to Multi-Parametric HIV-1 Genetics Using Multiple Displacement Amplification: Determining the What, How, and Where of the HIV-1 Reservoir.Viruses. 2021 Dec 10;13(12):2475. doi: 10.3390/v13122475. Viruses. 2021. PMID: 34960744 Free PMC article. Review.
-
CD161+ CD4+ T Cells Harbor Clonally Expanded Replication-Competent HIV-1 in Antiretroviral Therapy-Suppressed Individuals.mBio. 2019 Oct 8;10(5):e02121-19. doi: 10.1128/mBio.02121-19. mBio. 2019. PMID: 31594817 Free PMC article.
-
Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy.Science. 1997 Nov 14;278(5341):1295-300. doi: 10.1126/science.278.5341.1295. Science. 1997. PMID: 9360927
-
Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection.Nature. 1997 May 8;387(6629):183-8. doi: 10.1038/387183a0. Nature. 1997. PMID: 9144289
Cited by
-
Measuring the latent reservoir for HIV-1: Quantification bias in near full-length genome sequencing methods.PLoS Pathog. 2022 Sep 8;18(9):e1010845. doi: 10.1371/journal.ppat.1010845. eCollection 2022 Sep. PLoS Pathog. 2022. PMID: 36074794 Free PMC article.
-
TGF-β blockade drives a transitional effector phenotype in T cells reversing SIV latency and decreasing SIV reservoirs in vivo.Nat Commun. 2024 Feb 14;15(1):1348. doi: 10.1038/s41467-024-45555-x. Nat Commun. 2024. PMID: 38355731 Free PMC article.
-
Immune Control of HIV.J Life Sci (Westlake Village). 2019 Jun;1(1):4-37. J Life Sci (Westlake Village). 2019. PMID: 31468033 Free PMC article.
-
Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration.Nat Commun. 2019 Sep 6;10(1):4059. doi: 10.1038/s41467-019-12046-3. Nat Commun. 2019. PMID: 31492853 Free PMC article.
-
Rapamycin limits CD4+ T cell proliferation in simian immunodeficiency virus-infected rhesus macaques on antiretroviral therapy.J Clin Invest. 2022 May 16;132(10):e156063. doi: 10.1172/JCI156063. J Clin Invest. 2022. PMID: 35316218 Free PMC article.
References
-
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings / International Conference on Intelligent Systems for Molecular Biology ; ISMB International Conference on Intelligent Systems for Molecular Biology. 1994;2:28–36. - PubMed
Publication types
MeSH terms
Grants and funding
- UL1 TR000043/TR/NCATS NIH HHS/United States
- UM1 AI100645/AI/NIAID NIH HHS/United States
- UM1 AI100663/AI/NIAID NIH HHS/United States
- T32 AI070084/AI/NIAID NIH HHS/United States
- P01 AI100148/AI/NIAID NIH HHS/United States
- P30 AI045008/AI/NIAID NIH HHS/United States
- R37 AI066998/AI/NIAID NIH HHS/United States
- UM1 AI069481/AI/NIAID NIH HHS/United States
- 8 UL1 TR000043/TR/NCATS NIH HHS/United States
- UM1AI100663/AI/NIAID NIH HHS/United States
- Howard Hughes Medical Institute/United States
- R37 AI 066998/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
