

Epstein-Barr virus and Burkitt lymphoma
- PMID: 25418195
- PMCID: PMC4308657
- DOI: 10.5732/cjc.014.10190
Epstein-Barr virus and Burkitt lymphoma
Abstract
In 1964, a new herpesvirus, Epstein-Barr virus (EBV), was discovered in cultured tumor cells derived from a Burkitt lymphoma (BL) biopsy taken from an African patient. This was a momentous event that reinvigorated research into viruses as a possible cause of human cancers. Subsequent studies demonstrated that EBV was a potent growth-transforming agent for primary B cells, and that all cases of BL carried characteristic chromosomal translocations resulting in constitutive activation of the c-MYC oncogene. These results hinted at simple oncogenic mechanisms that would make Burkitt lymphoma paradigmatic for cancers with viral etiology. In reality, the pathogenesis of this tumor is rather complicated with regard to both the contribution of the virus and the involvement of cellular oncogenes. Here, we review the current understanding of the roles of EBV and c-MYC in the pathogenesis of BL and the implications for new therapeutic strategies to treat this lymphoma.
Figures
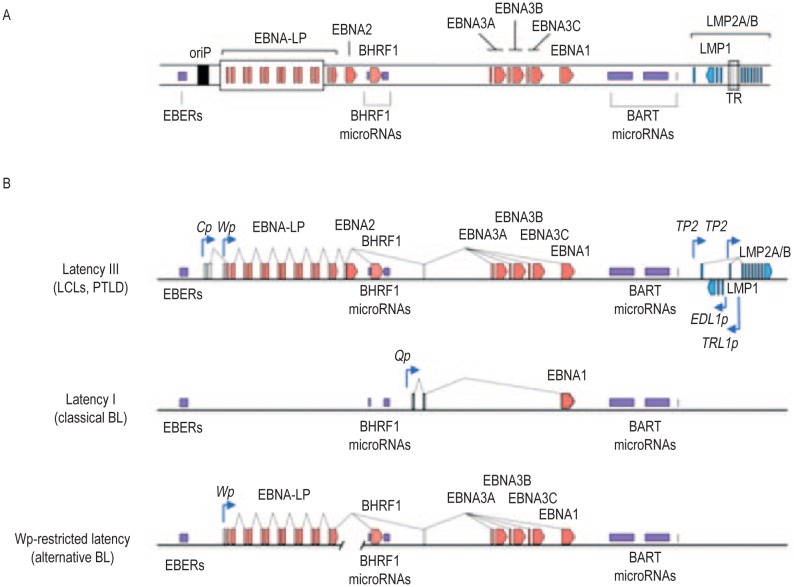
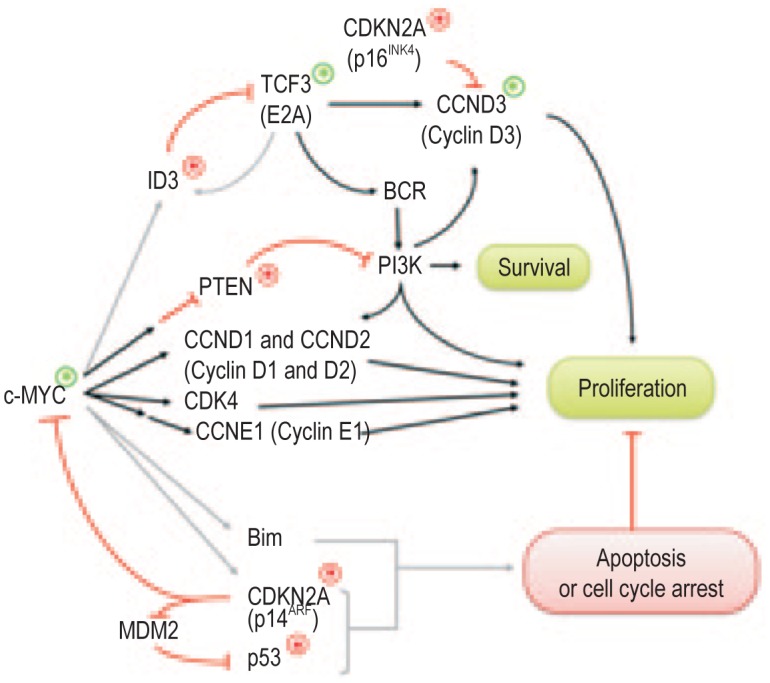
Similar articles
-
Epstein-Barr virus and the pathogenesis of Burkitt's lymphoma: more questions than answers.Int J Cancer. 2009 Apr 15;124(8):1745-55. doi: 10.1002/ijc.24223. Int J Cancer. 2009. PMID: 19165855 Review.
-
Epstein-Barr virus and Burkitt lymphoma.J Clin Pathol. 2007 Dec;60(12):1397-402. doi: 10.1136/jcp.2007.047977. J Clin Pathol. 2007. PMID: 18042696 Free PMC article. Review.
-
MYC directly transactivates CR2/CD21, the receptor of the Epstein-Barr virus, enhancing the viral infection of Burkitt lymphoma cells.Oncogene. 2023 Nov;42(45):3358-3370. doi: 10.1038/s41388-023-02846-9. Epub 2023 Sep 29. Oncogene. 2023. PMID: 37773203
-
Three restricted forms of Epstein-Barr virus latency counteracting apoptosis in c-myc-expressing Burkitt lymphoma cells.Proc Natl Acad Sci U S A. 2006 Oct 3;103(40):14935-40. doi: 10.1073/pnas.0509988103. Epub 2006 Sep 25. Proc Natl Acad Sci U S A. 2006. PMID: 17001014 Free PMC article.
-
Burkitt lymphoma: revisiting the pathogenesis of a virus-associated malignancy.Hematology Am Soc Hematol Educ Program. 2007:277-84. doi: 10.1182/asheducation-2007.1.277. Hematology Am Soc Hematol Educ Program. 2007. PMID: 18024641 Review.
Cited by
-
Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103.Cell Death Dis. 2016 Sep 1;7(9):e2353. doi: 10.1038/cddis.2016.253. Cell Death Dis. 2016. PMID: 27584792 Free PMC article.
-
Case-control study of paternal occupational exposures and childhood lymphoma in Great Britain, 1962-2010.Br J Cancer. 2019 Jun;120(12):1153-1161. doi: 10.1038/s41416-019-0469-7. Epub 2019 May 20. Br J Cancer. 2019. PMID: 31105271 Free PMC article.
-
FAMLF is a target of miR-181b in Burkitt lymphoma.Braz J Med Biol Res. 2017 May 4;50(6):e5661. doi: 10.1590/1414-431X20175661. Braz J Med Biol Res. 2017. PMID: 28492808 Free PMC article.
-
The oncogenic role of Epstein-Barr virus-encoded microRNAs in Epstein-Barr virus-associated gastric carcinoma.J Cell Mol Med. 2018 Jan;22(1):38-45. doi: 10.1111/jcmm.13354. Epub 2017 Oct 9. J Cell Mol Med. 2018. PMID: 28990284 Free PMC article. Review.
-
Design of a multi-epitopic vaccine against Epstein-Barr virus via computer-based methods.Front Immunol. 2023 Mar 14;14:1115345. doi: 10.3389/fimmu.2023.1115345. eCollection 2023. Front Immunol. 2023. PMID: 36999015 Free PMC article.
References
-
- De-The G. The epidemiology of Burkitt's lymphoma: evidence for a causal association with Epstein-Barr virus. Epidemiol Rev. 1979;1:32–54. - PubMed
-
- Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156:744–756. - PubMed
-
- Henle G, Henle W, Clifford P, et al. Antibodies to Epstein-Barr virus in Burkitt's lymphoma and control groups. J Natl Cancer Inst. 1969;43:1147–1157. - PubMed
-
- Klein G, Clifford P, Henle G, et al. EBV-associated serological patterns in a Burkitt lymphoma patient during regression and recurrence. Int J Cancer. 1969;4:416–421. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources