

Synchronized renal tubular cell death involves ferroptosis
- PMID: 25385600
- PMCID: PMC4250130
- DOI: 10.1073/pnas.1415518111
Synchronized renal tubular cell death involves ferroptosis
Abstract
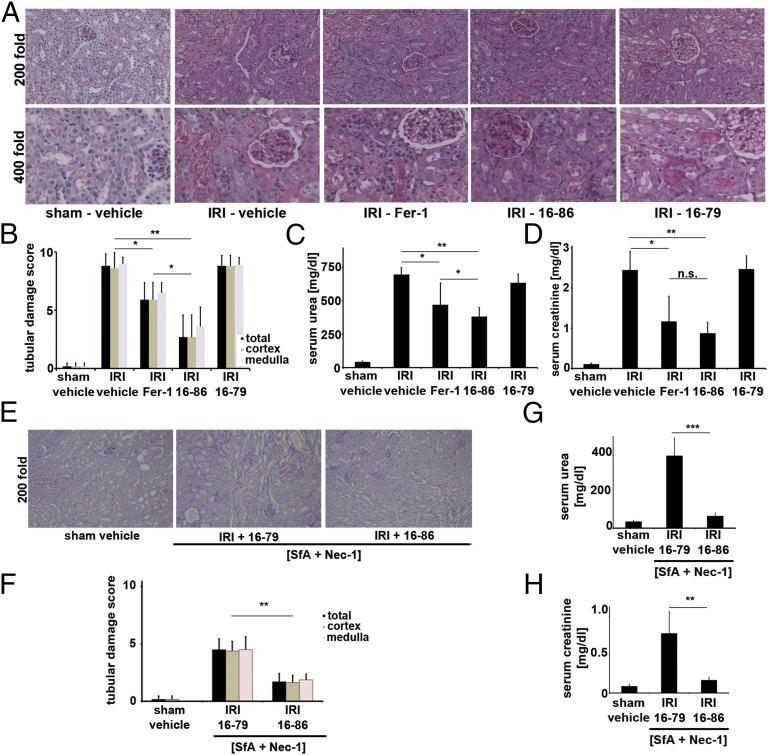
Receptor-interacting protein kinase 3 (RIPK3)-mediated necroptosis is thought to be the pathophysiologically predominant pathway that leads to regulated necrosis of parenchymal cells in ischemia-reperfusion injury (IRI), and loss of either Fas-associated protein with death domain (FADD) or caspase-8 is known to sensitize tissues to undergo spontaneous necroptosis. Here, we demonstrate that renal tubules do not undergo sensitization to necroptosis upon genetic ablation of either FADD or caspase-8 and that the RIPK1 inhibitor necrostatin-1 (Nec-1) does not protect freshly isolated tubules from hypoxic injury. In contrast, iron-dependent ferroptosis directly causes synchronized necrosis of renal tubules, as demonstrated by intravital microscopy in models of IRI and oxalate crystal-induced acute kidney injury. To suppress ferroptosis in vivo, we generated a novel third-generation ferrostatin (termed 16-86), which we demonstrate to be more stable, to metabolism and plasma, and more potent, compared with the first-in-class compound ferrostatin-1 (Fer-1). Even in conditions with extraordinarily severe IRI, 16-86 exerts strong protection to an extent which has not previously allowed survival in any murine setting. In addition, 16-86 further potentiates the strong protective effect on IRI mediated by combination therapy with necrostatins and compounds that inhibit mitochondrial permeability transition. Renal tubules thus represent a tissue that is not sensitized to necroptosis by loss of FADD or caspase-8. Finally, ferroptosis mediates postischemic and toxic renal necrosis, which may be therapeutically targeted by ferrostatins and by combination therapy.
Keywords: apoptosis; ferroptosis; necroptosis; programmed cell death; regulated cell death.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Loss of receptor interacting protein kinases 3 and caspase-8 augments intrinsic apoptosis in tubular epithelial cell and promote kidney ischaemia-reperfusion injury.Nephrology (Carlton). 2019 Jun;24(6):661-669. doi: 10.1111/nep.13487. Epub 2019 Apr 29. Nephrology (Carlton). 2019. PMID: 30175514 Free PMC article.
-
Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury.Kidney Int. 2012 Apr;81(8):751-61. doi: 10.1038/ki.2011.450. Epub 2012 Jan 11. Kidney Int. 2012. PMID: 22237751
-
TWEAK and RIPK1 mediate a second wave of cell death during AKI.Proc Natl Acad Sci U S A. 2018 Apr 17;115(16):4182-4187. doi: 10.1073/pnas.1716578115. Epub 2018 Mar 27. Proc Natl Acad Sci U S A. 2018. PMID: 29588419 Free PMC article.
-
Regulated necrosis in kidney ischemia-reperfusion injury.Kidney Int. 2019 Aug;96(2):291-301. doi: 10.1016/j.kint.2019.02.009. Epub 2019 Mar 7. Kidney Int. 2019. PMID: 31005270 Review.
-
Developmental checkpoints guarded by regulated necrosis.Cell Mol Life Sci. 2016 Jun;73(11-12):2125-36. doi: 10.1007/s00018-016-2188-z. Epub 2016 Apr 7. Cell Mol Life Sci. 2016. PMID: 27056574 Free PMC article. Review.
Cited by
-
Lipid peroxidation and the subsequent cell death transmitting from ferroptotic cells to neighboring cells.Cell Death Dis. 2021 Mar 29;12(4):332. doi: 10.1038/s41419-021-03613-y. Cell Death Dis. 2021. PMID: 33782392 Free PMC article.
-
Waves of ferroptotic cell death sculpt embryonic tissue.Nature. 2024 Jul;631(8021):510-512. doi: 10.1038/d41586-024-02125-x. Nature. 2024. PMID: 38987333 No abstract available.
-
Epigenetic Regulation of Ferroptosis in the Liver.Research (Wash D C). 2024 Feb 21;7:0323. doi: 10.34133/research.0323. eCollection 2024. Research (Wash D C). 2024. PMID: 38384329 Free PMC article. Review. No abstract available.
-
Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury.Antioxidants (Basel). 2021 Apr 25;10(5):667. doi: 10.3390/antiox10050667. Antioxidants (Basel). 2021. PMID: 33922912 Free PMC article. Review.
-
Primary cilia suppress Ripk3-mediated necroptosis.Cell Death Discov. 2022 Dec 2;8(1):477. doi: 10.1038/s41420-022-01272-2. Cell Death Discov. 2022. PMID: 36460631 Free PMC article.
References
-
- Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35C:24–32. - PubMed
-
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–223. - PubMed
-
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
