

IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications
- PMID: 25375629
- PMCID: PMC4223080
- DOI: 10.1371/journal.ppat.1004503
IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications
Erratum in
-
Correction: IFI16 Restricts HSV-1 Replication by Accumulating on the HSV-1 Genome, Repressing HSV-1 Gene Expression, and Directly or Indirectly Modulating Histone Modifications.PLoS Pathog. 2018 Jun 6;14(6):e1007113. doi: 10.1371/journal.ppat.1007113. eCollection 2018 Jun. PLoS Pathog. 2018. PMID: 29874275 Free PMC article.
Abstract
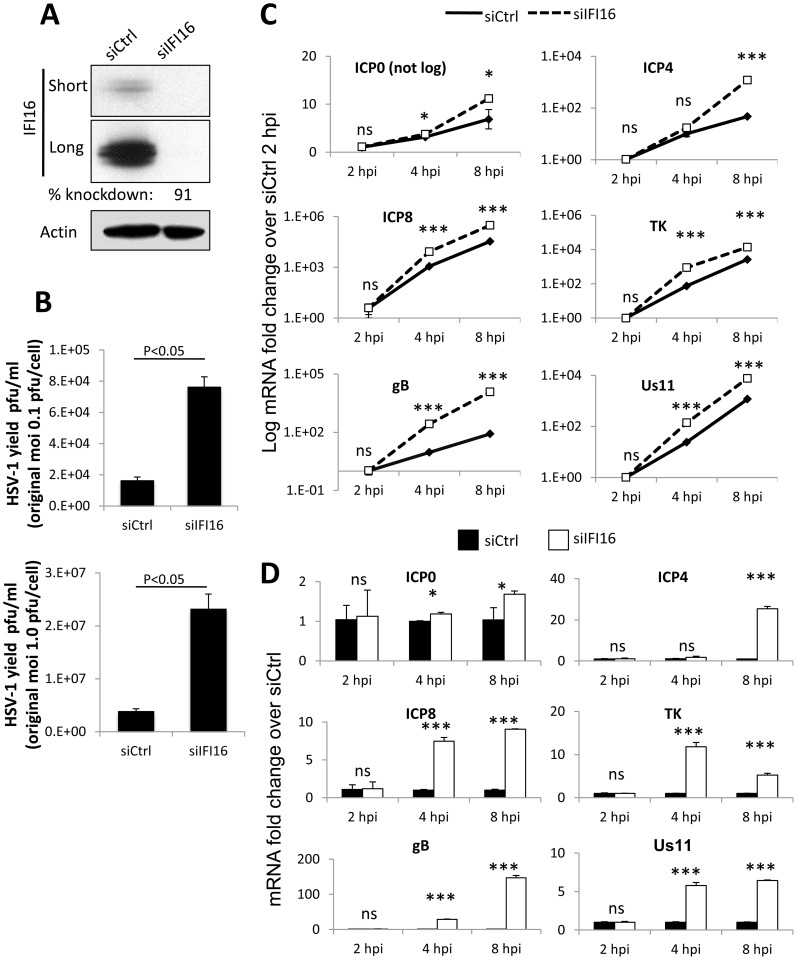
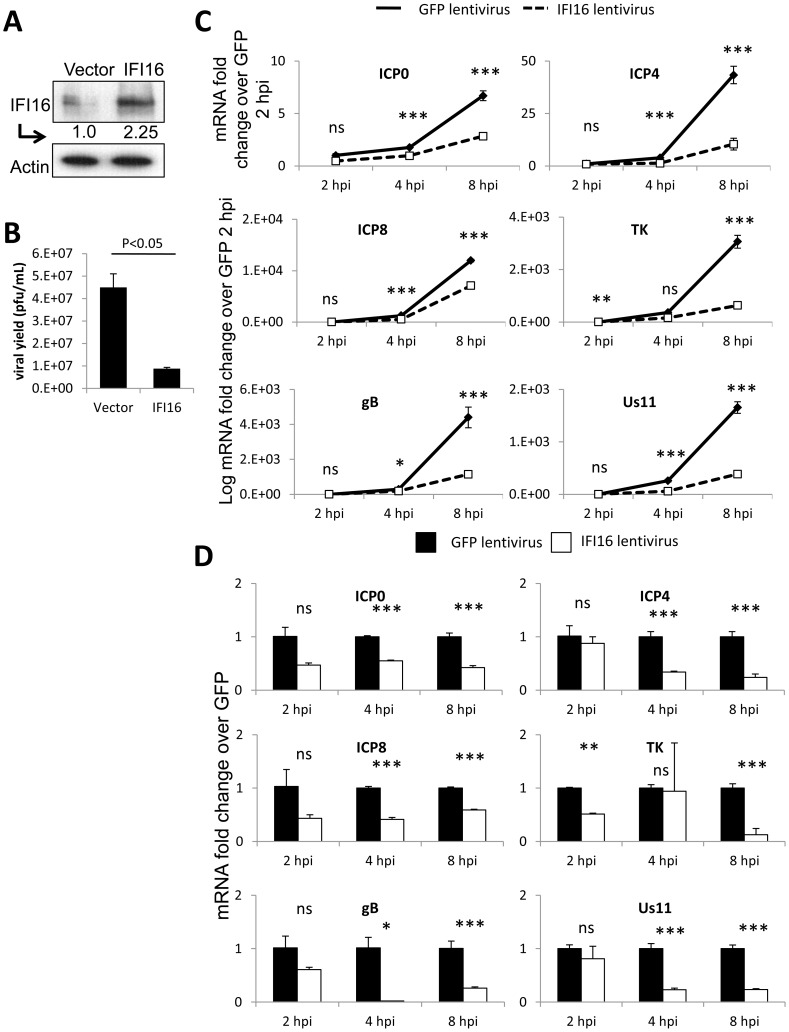
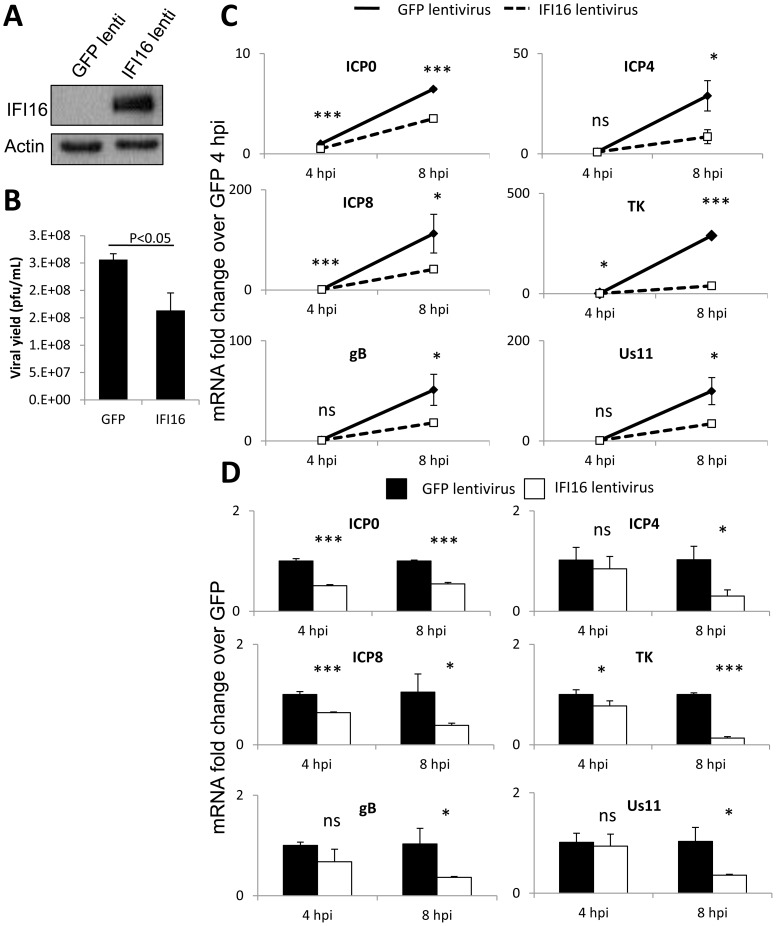
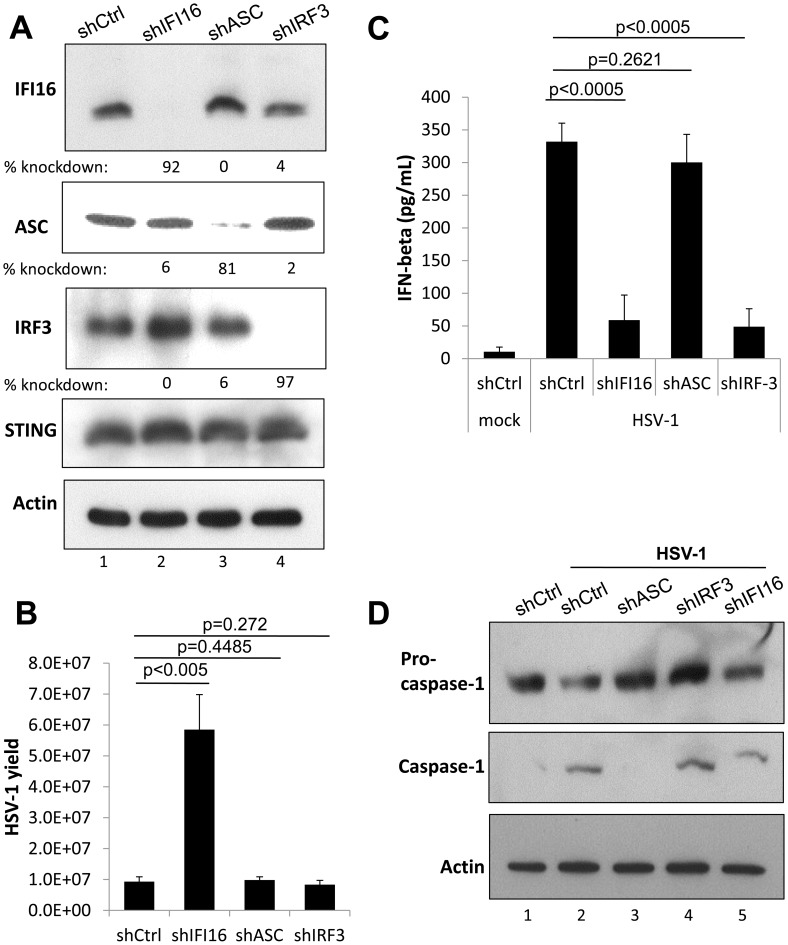
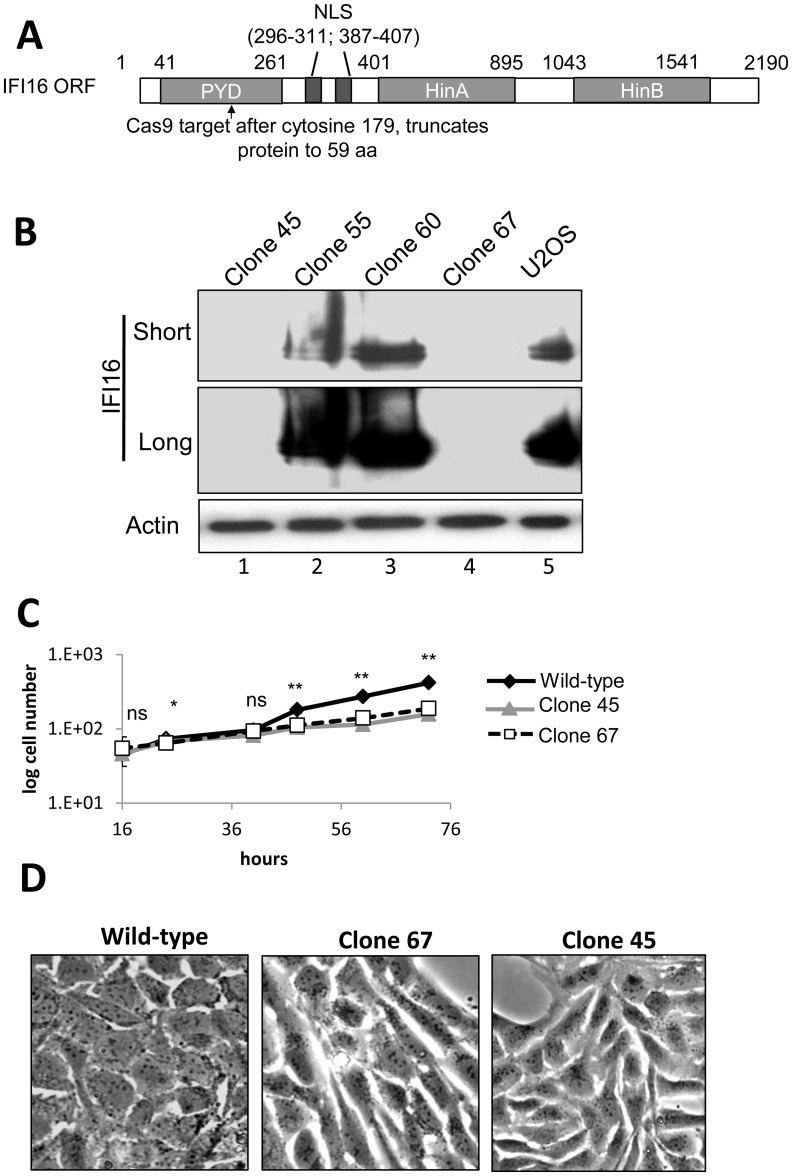
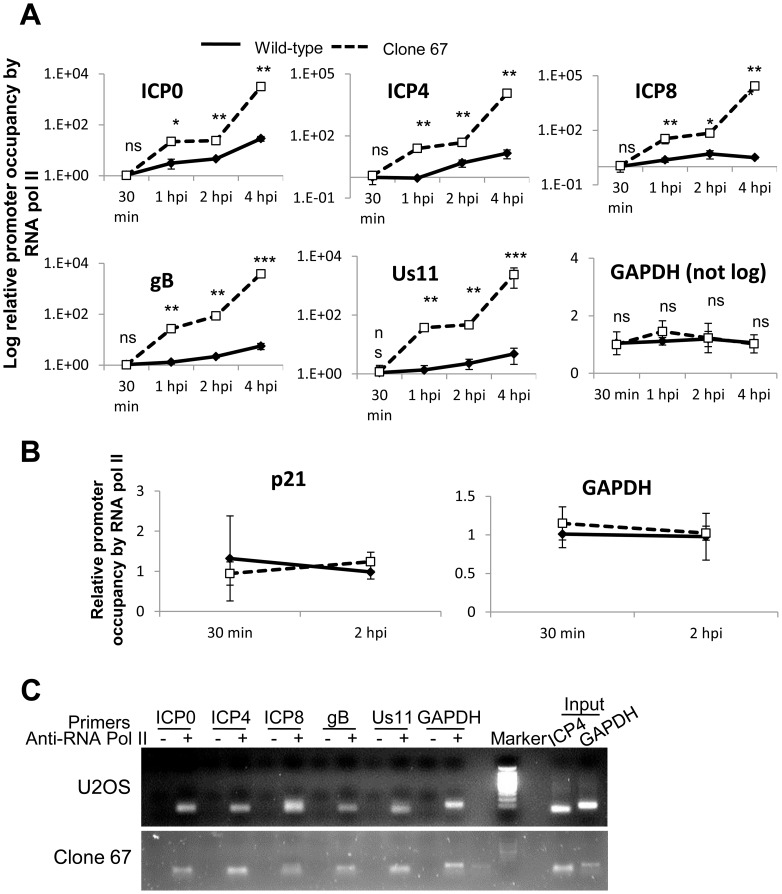
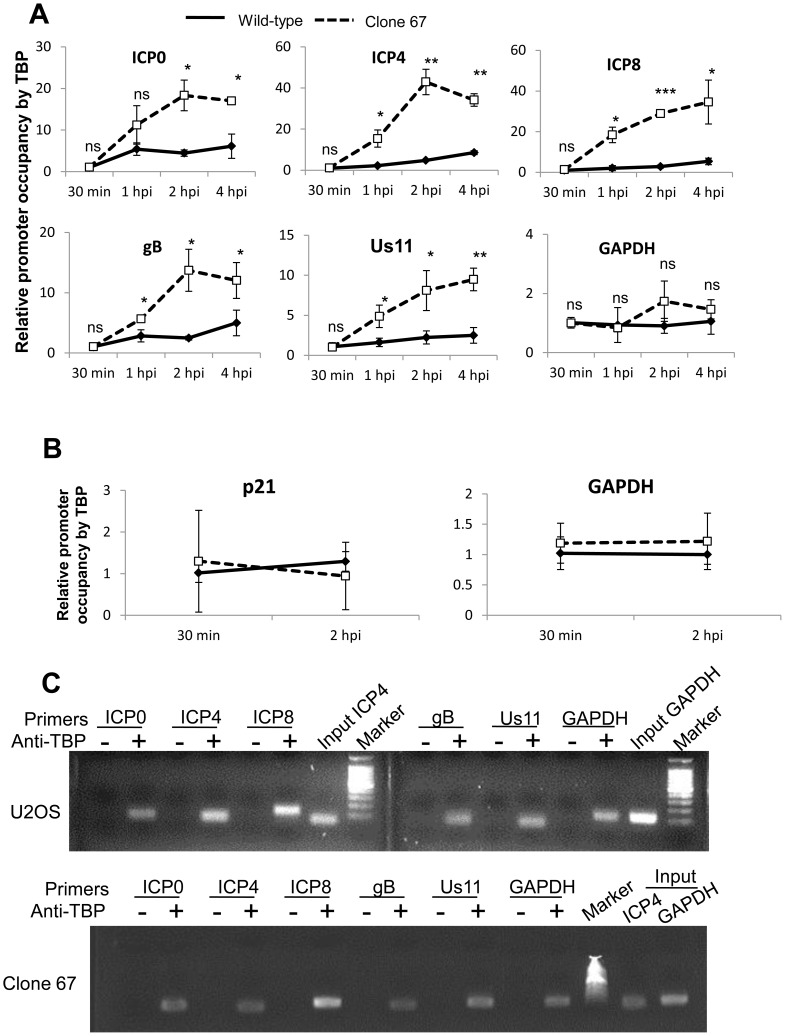
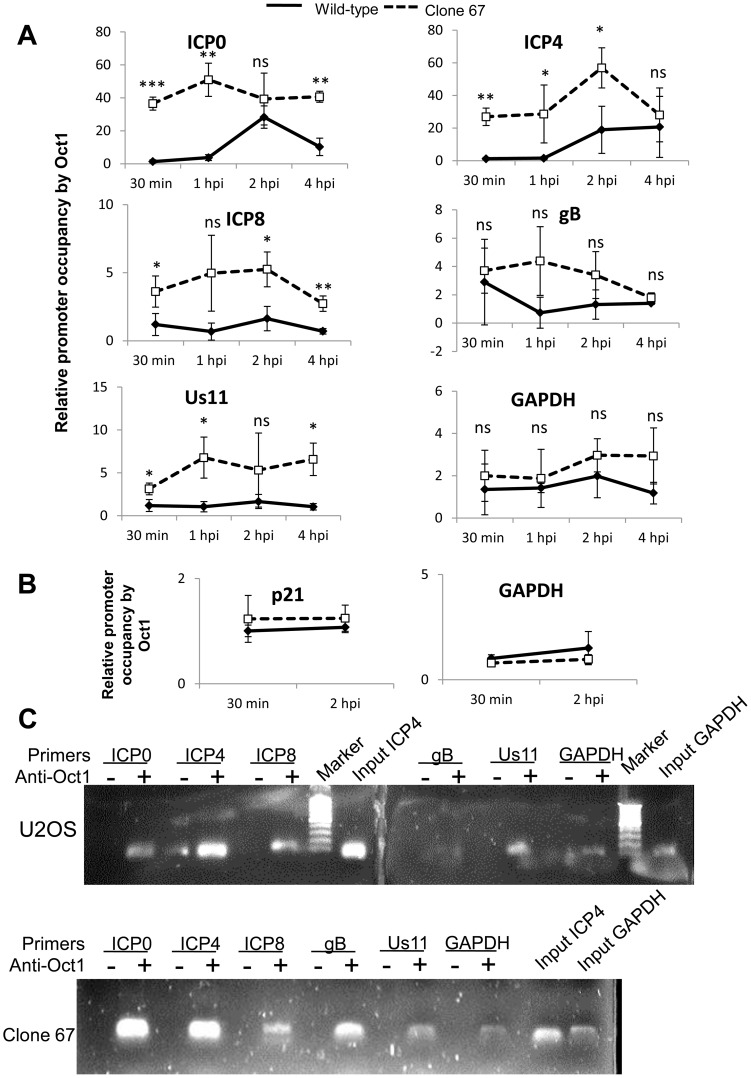
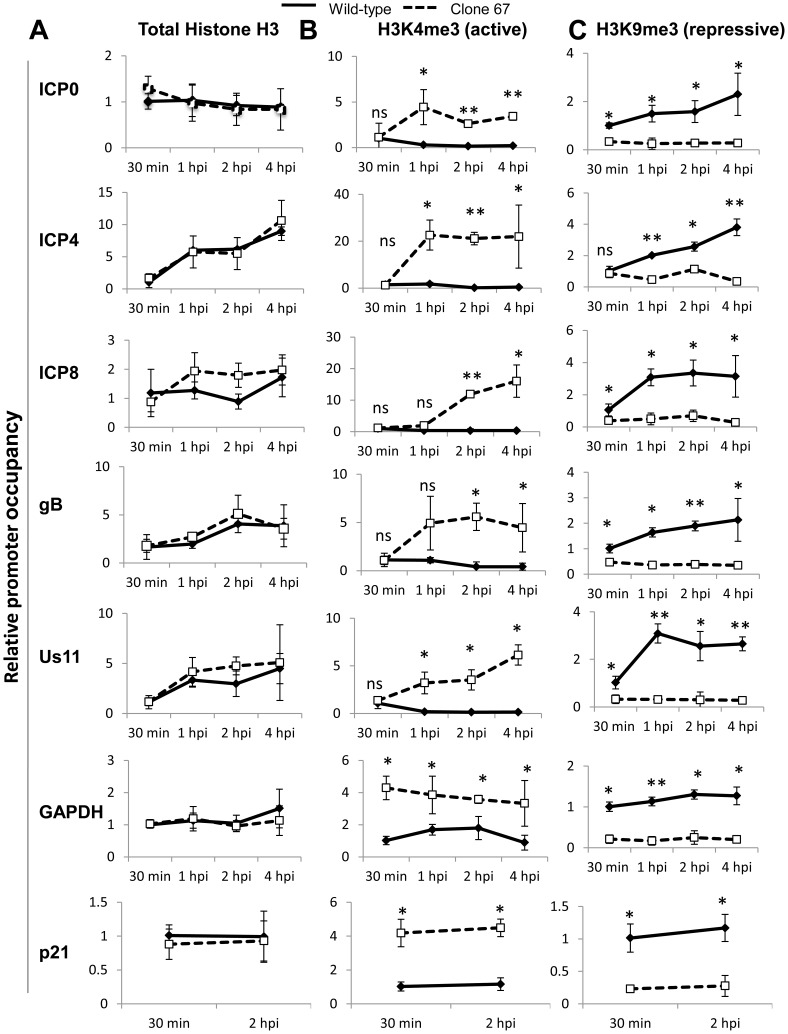
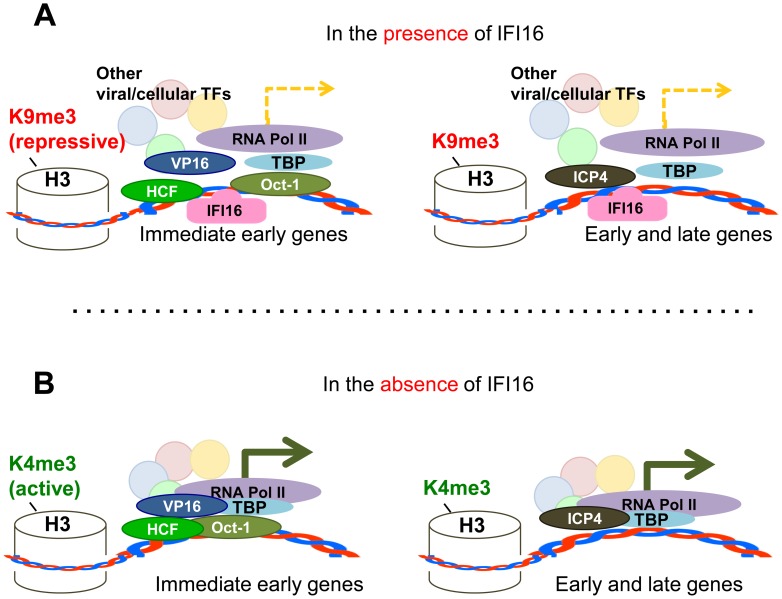
Interferon-γ inducible factor 16 (IFI16) is a multifunctional nuclear protein involved in transcriptional regulation, induction of interferon-β (IFN-β), and activation of the inflammasome response. It interacts with the sugar-phosphate backbone of dsDNA and modulates viral and cellular transcription through largely undetermined mechanisms. IFI16 is a restriction factor for human cytomegalovirus (HCMV) and herpes simplex virus (HSV-1), though the mechanisms of HSV-1 restriction are not yet understood. Here, we show that IFI16 has a profound effect on HSV-1 replication in human foreskin fibroblasts, osteosarcoma cells, and breast epithelial cancer cells. IFI16 knockdown increased HSV-1 yield 6-fold and IFI16 overexpression reduced viral yield by over 5-fold. Importantly, HSV-1 gene expression, including the immediate early proteins, ICP0 and ICP4, the early proteins, ICP8 and TK, and the late proteins gB and Us11, was reduced in the presence of IFI16. Depletion of the inflammasome adaptor protein, ASC, or the IFN-inducing transcription factor, IRF-3, did not affect viral yield. ChIP studies demonstrated the presence of IFI16 bound to HSV-1 promoters in osteosarcoma (U2OS) cells and fibroblasts. Using CRISPR gene editing technology, we generated U2OS cells with permanent deletion of IFI16 protein expression. ChIP analysis of these cells and wild-type (wt) U2OS demonstrated increased association of RNA polymerase II, TATA binding protein (TBP) and Oct1 transcription factors with viral promoters in the absence of IFI16 at different times post infection. Although IFI16 did not alter the total histone occupancy at viral or cellular promoters, its absence promoted markers of active chromatin and decreased those of repressive chromatin with viral and cellular gene promoters. Collectively, these studies for the first time demonstrate that IFI16 prevents association of important transcriptional activators with wt HSV-1 promoters and suggest potential mechanisms of IFI16 restriction of wt HSV-1 replication and a direct or indirect role for IFI16 in histone modification.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



Similar articles
-
Mechanisms of Host IFI16, PML, and Daxx Protein Restriction of Herpes Simplex Virus 1 Replication.J Virol. 2018 Apr 27;92(10):e00057-18. doi: 10.1128/JVI.00057-18. Print 2018 May 15. J Virol. 2018. PMID: 29491153 Free PMC article.
-
The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters.J Virol. 2015 Aug;89(15):7506-20. doi: 10.1128/JVI.00013-15. Epub 2015 May 13. J Virol. 2015. PMID: 25972554 Free PMC article.
-
Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi's Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency.J Virol. 2016 Sep 12;90(19):8822-41. doi: 10.1128/JVI.01003-16. Print 2016 Oct 1. J Virol. 2016. PMID: 27466416 Free PMC article.
-
Role of ND10 nuclear bodies in the chromatin repression of HSV-1.Virol J. 2016 Apr 5;13:62. doi: 10.1186/s12985-016-0516-4. Virol J. 2016. PMID: 27048561 Free PMC article. Review.
-
Nuclear sensing of viral DNA, epigenetic regulation of herpes simplex virus infection, and innate immunity.Virology. 2015 May;479-480:153-9. doi: 10.1016/j.virol.2015.02.009. Epub 2015 Mar 3. Virology. 2015. PMID: 25742715 Free PMC article. Review.
Cited by
-
Herpesvirus Genome Recognition Induced Acetylation of Nuclear IFI16 Is Essential for Its Cytoplasmic Translocation, Inflammasome and IFN-β Responses.PLoS Pathog. 2015 Jul 2;11(7):e1005019. doi: 10.1371/journal.ppat.1005019. eCollection 2015 Jul. PLoS Pathog. 2015. PMID: 26134128 Free PMC article.
-
Immune Sensing Mechanisms that Discriminate Self from Altered Self and Foreign Nucleic Acids.Immunity. 2020 Jul 14;53(1):54-77. doi: 10.1016/j.immuni.2020.06.014. Immunity. 2020. PMID: 32668228 Free PMC article. Review.
-
HSV-1 exploits host heterochromatin for nuclear egress.J Cell Biol. 2023 Sep 4;222(9):e202304106. doi: 10.1083/jcb.202304106. Epub 2023 Jul 26. J Cell Biol. 2023. PMID: 37516914 Free PMC article.
-
Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis.Pathogens. 2023 Mar 10;12(3):437. doi: 10.3390/pathogens12030437. Pathogens. 2023. PMID: 36986359 Free PMC article. Review.
-
The Telomeric Response to Viral Infection.Viruses. 2017 Aug 9;9(8):218. doi: 10.3390/v9080218. Viruses. 2017. PMID: 28792463 Free PMC article. Review.
References
-
- Knipe DM, Howley PM, editors (2006) Fields' Virology. Philadelphia: Lippincott Williams & Wilkins. 3177 p.
-
- Streilein JW, Dana MR, Ksander BR (1997) Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol Today 18: 443–449. - PubMed
-
- Whitley RJ, Gnann JW (2002) Viral encephalitis: familiar infections and emerging pathogens. Lancet 359: 507–513. - PubMed
-
- Whitley RJ, Soong SJ, Linneman C Jr, Liu C, Pazin G, et al. (1982) Herpes simplex encephalitis. Clinical Assessment. JAMA 247: 317–320. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
