

HTSeq--a Python framework to work with high-throughput sequencing data
- PMID: 25260700
- PMCID: PMC4287950
- DOI: 10.1093/bioinformatics/btu638
HTSeq--a Python framework to work with high-throughput sequencing data
Abstract
Motivation: A large choice of tools exists for many standard tasks in the analysis of high-throughput sequencing (HTS) data. However, once a project deviates from standard workflows, custom scripts are needed.
Results: We present HTSeq, a Python library to facilitate the rapid development of such scripts. HTSeq offers parsers for many common data formats in HTS projects, as well as classes to represent data, such as genomic coordinates, sequences, sequencing reads, alignments, gene model information and variant calls, and provides data structures that allow for querying via genomic coordinates. We also present htseq-count, a tool developed with HTSeq that preprocesses RNA-Seq data for differential expression analysis by counting the overlap of reads with genes.
Availability and implementation: HTSeq is released as an open-source software under the GNU General Public Licence and available from http://www-huber.embl.de/HTSeq or from the Python Package Index at https://pypi.python.org/pypi/HTSeq.
© The Author 2014. Published by Oxford University Press.
Figures
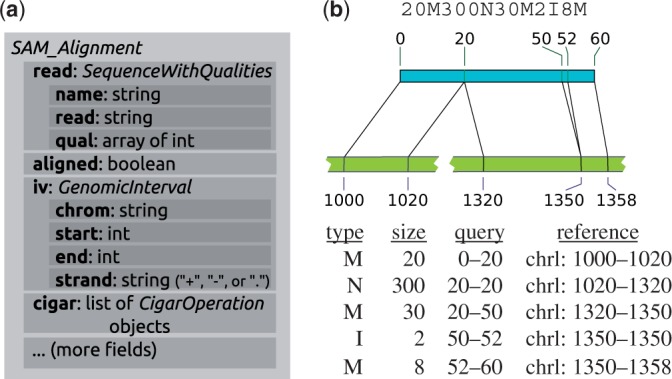
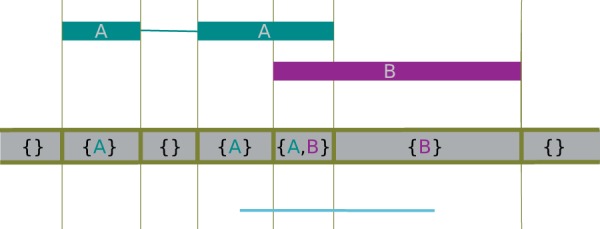
Similar articles
-
Analysing high-throughput sequencing data in Python with HTSeq 2.0.Bioinformatics. 2022 May 13;38(10):2943-2945. doi: 10.1093/bioinformatics/btac166. Bioinformatics. 2022. PMID: 35561197 Free PMC article.
-
htseq-clip: a toolset for the preprocessing of eCLIP/iCLIP datasets.Bioinformatics. 2023 Jan 1;39(1):btac747. doi: 10.1093/bioinformatics/btac747. Bioinformatics. 2023. PMID: 36394253 Free PMC article.
-
Rcount: simple and flexible RNA-Seq read counting.Bioinformatics. 2015 Feb 1;31(3):436-7. doi: 10.1093/bioinformatics/btu680. Epub 2014 Oct 15. Bioinformatics. 2015. PMID: 25322836
-
Omics Pipe: a community-based framework for reproducible multi-omics data analysis.Bioinformatics. 2015 Jun 1;31(11):1724-8. doi: 10.1093/bioinformatics/btv061. Epub 2015 Jan 30. Bioinformatics. 2015. PMID: 25637560 Free PMC article.
-
Bioinformatics tools for analysing viral genomic data.Rev Sci Tech. 2016 Apr;35(1):271-85. doi: 10.20506/rst.35.1.2432. Rev Sci Tech. 2016. PMID: 27217183 Review.
Cited by
-
Perturbations in eIF3 subunit stoichiometry alter expression of ribosomal proteins and key components of the MAPK signaling pathways.Elife. 2024 Nov 4;13:RP95846. doi: 10.7554/eLife.95846. Elife. 2024. PMID: 39495207 Free PMC article.
-
Conversion of glioma cells into neuron-like cells by small molecules.iScience. 2024 Oct 2;27(11):111091. doi: 10.1016/j.isci.2024.111091. eCollection 2024 Nov 15. iScience. 2024. PMID: 39483145 Free PMC article.
-
Integration of fungal transcriptomics and metabolomics provides insights into the early interaction between the ORM fungus Tulasnella sp. and the orchid Serapias vomeracea seeds.IMA Fungus. 2024 Oct 25;15(1):31. doi: 10.1186/s43008-024-00165-6. IMA Fungus. 2024. PMID: 39456087 Free PMC article.
-
Maize unstable factor for orange1 is essential for endosperm development and carbohydrate accumulation.Plant Physiol. 2021 Aug 3;186(4):1932-1950. doi: 10.1093/plphys/kiab183. Plant Physiol. 2021. PMID: 33905500 Free PMC article.
-
Crohn's disease-associated ATG16L1 T300A genotype is associated with improved survival in gastric cancer.EBioMedicine. 2021 May;67:103347. doi: 10.1016/j.ebiom.2021.103347. Epub 2021 Apr 25. EBioMedicine. 2021. PMID: 33906066 Free PMC article.
References
-
- Beazley DM, et al. Proceedings of the 4th USENIX Tcl/Tk workshop. 1996. SWIG: an easy to use tool for integrating scripting languages with C and C++ pp. 129–139.
-
- Behnel S, et al. Cython: the best of both worlds. Comput. Sci. Eng. 2011;13:31–39.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
