

CCMpred--fast and precise prediction of protein residue-residue contacts from correlated mutations
- PMID: 25064567
- PMCID: PMC4201158
- DOI: 10.1093/bioinformatics/btu500
CCMpred--fast and precise prediction of protein residue-residue contacts from correlated mutations
Abstract
Motivation: Recent breakthroughs in protein residue-residue contact prediction have made reliable de novo prediction of protein structures possible. The key was to apply statistical methods that can distinguish direct couplings between pairs of columns in a multiple sequence alignment from merely correlated pairs, i.e. to separate direct from indirect effects. Two classes of such methods exist, either relying on regularized inversion of the covariance matrix or on pseudo-likelihood maximization (PLM). Although PLM-based methods offer clearly higher precision, available tools are not sufficiently optimized and are written in interpreted languages that introduce additional overheads. This impedes the runtime and large-scale contact prediction for larger protein families, multi-domain proteins and protein-protein interactions.
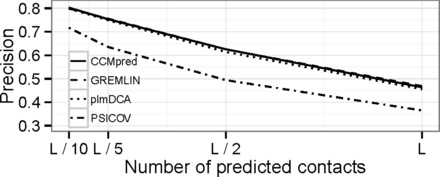
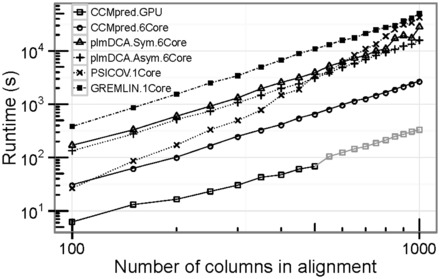
Results: Here we introduce CCMpred, our performance-optimized PLM implementation in C and CUDA C. Using graphics cards in the price range of current six-core processors, CCMpred can predict contacts for typical alignments 35-113 times faster and with the same precision as the most accurate published methods. For users without a CUDA-capable graphics card, CCMpred can also run in a CPU mode that is still 4-14 times faster. Thanks to our speed-ups (http://dictionary.cambridge.org/dictionary/british/speed-up) contacts for typical protein families can be predicted in 15-60 s on a consumer-grade GPU and 1-6 min on a six-core CPU.
Availability and implementation: CCMpred is free and open-source software under the GNU Affero General Public License v3 (or later) available at https://bitbucket.org/soedinglab/ccmpred.
© The Author 2014. Published by Oxford University Press.
Figures
Similar articles
-
MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins.Bioinformatics. 2015 Apr 1;31(7):999-1006. doi: 10.1093/bioinformatics/btu791. Epub 2014 Nov 26. Bioinformatics. 2015. PMID: 25431331 Free PMC article.
-
bbcontacts: prediction of β-strand pairing from direct coupling patterns.Bioinformatics. 2015 Jun 1;31(11):1729-37. doi: 10.1093/bioinformatics/btv041. Epub 2015 Jan 23. Bioinformatics. 2015. PMID: 25618863
-
The evolution of contact prediction: evidence that contact selection in statistical contact prediction is changing.Bioinformatics. 2020 Mar 1;36(6):1750-1756. doi: 10.1093/bioinformatics/btz816. Bioinformatics. 2020. PMID: 31693112
-
High precision in protein contact prediction using fully convolutional neural networks and minimal sequence features.Bioinformatics. 2018 Oct 1;34(19):3308-3315. doi: 10.1093/bioinformatics/bty341. Bioinformatics. 2018. PMID: 29718112 Free PMC article.
-
Predicting accurate contacts in thousands of Pfam domain families using PconsC3.Bioinformatics. 2017 Sep 15;33(18):2859-2866. doi: 10.1093/bioinformatics/btx332. Bioinformatics. 2017. PMID: 28535189
Cited by
-
Residue contacts predicted by evolutionary covariance extend the application of ab initio molecular replacement to larger and more challenging protein folds.IUCrJ. 2016 Jun 15;3(Pt 4):259-70. doi: 10.1107/S2052252516008113. eCollection 2016 Jul 1. IUCrJ. 2016. PMID: 27437113 Free PMC article.
-
Ensemble Learning with Supervised Methods Based on Large-Scale Protein Language Models for Protein Mutation Effects Prediction.Int J Mol Sci. 2023 Nov 18;24(22):16496. doi: 10.3390/ijms242216496. Int J Mol Sci. 2023. PMID: 38003686 Free PMC article.
-
DeepTM: A deep learning algorithm for prediction of melting temperature of thermophilic proteins directly from sequences.Comput Struct Biotechnol J. 2023 Nov 4;21:5544-5560. doi: 10.1016/j.csbj.2023.11.006. eCollection 2023. Comput Struct Biotechnol J. 2023. PMID: 38034401 Free PMC article.
-
CopulaNet: Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction.Nat Commun. 2021 May 5;12(1):2535. doi: 10.1038/s41467-021-22869-8. Nat Commun. 2021. PMID: 33953201 Free PMC article.
-
SESNet: sequence-structure feature-integrated deep learning method for data-efficient protein engineering.J Cheminform. 2023 Feb 3;15(1):12. doi: 10.1186/s13321-023-00688-x. J Cheminform. 2023. PMID: 36737798 Free PMC article.
References
-
- Dunn SD, et al. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction. Bioinformatics. 2008;24:333–340. - PubMed
-
- Ekeberg M, et al. Improved contact prediction in proteins: using pseudolikelihoods to infer potts models. Phys. Rev. E. 2013;87:012707. - PubMed
-
- Jones DT, et al. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28:184–190. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
