

Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China
- PMID: 24719429
- PMCID: PMC4054348
- DOI: 10.1128/JVI.00631-14
Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China
Abstract
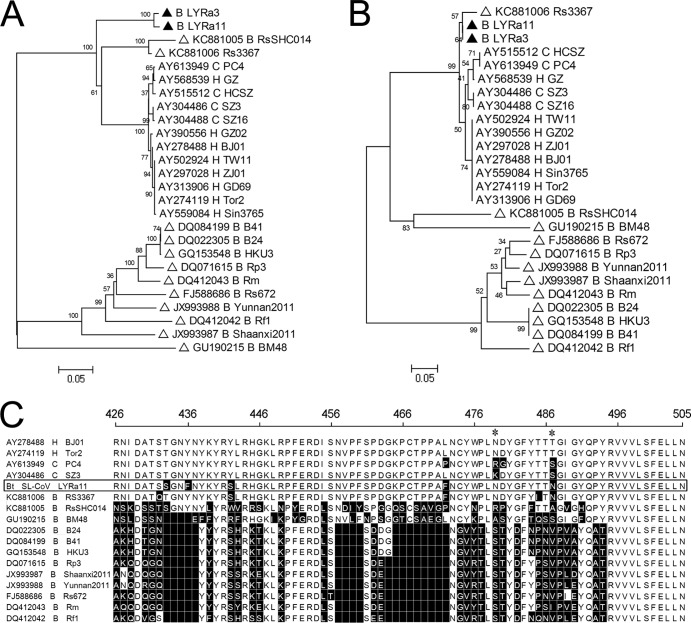
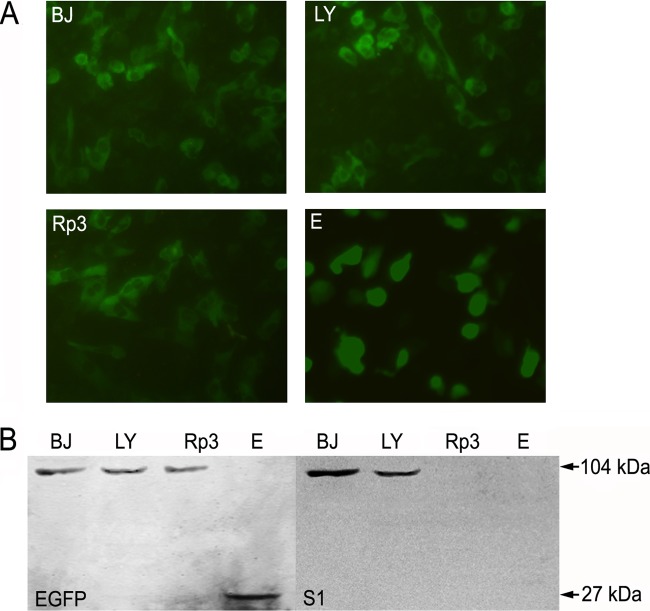
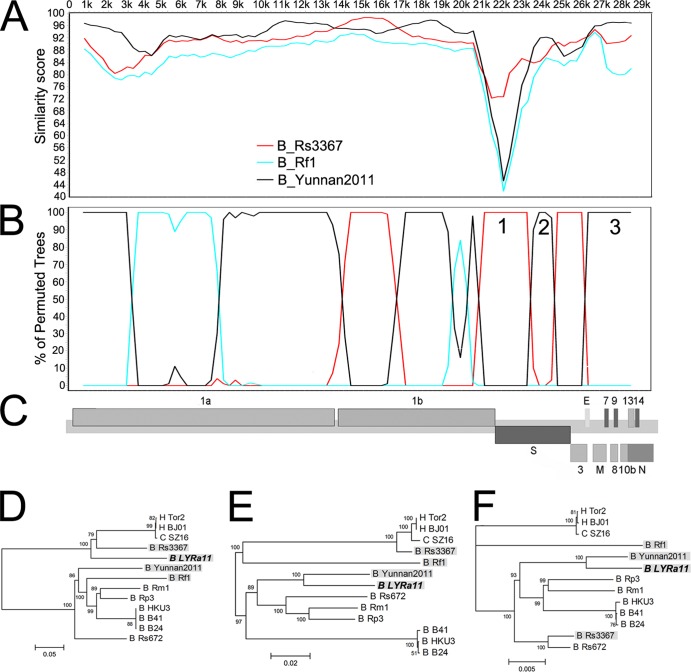
Although many severe acute respiratory syndrome-like coronaviruses (SARS-like CoVs) have been identified in bats in China, Europe, and Africa, most have a genetic organization significantly distinct from human/civet SARS CoVs in the receptor-binding domain (RBD), which mediates receptor binding and determines the host spectrum, resulting in their failure to cause human infections and making them unlikely progenitors of human/civet SARS CoVs. Here, a viral metagenomic analysis of 268 bat rectal swabs collected from four counties in Yunnan Province has identified hundreds of sequences relating to alpha- and betacoronaviruses. Phylogenetic analysis based on a conserved region of the RNA-dependent RNA polymerase gene revealed that alphacoronaviruses had diversities with some obvious differences from those reported previously. Full genomic analysis of a new SARS-like CoV from Baoshan (LYRa11) showed that it was 29,805 nucleotides (nt) in length with 13 open reading frames (ORFs), sharing 91% nucleotide identity with human/civet SARS CoVs and the most recently reported SARS-like CoV Rs3367, while sharing 89% with other bat SARS-like CoVs. Notably, it showed the highest sequence identity with the S gene of SARS CoVs and Rs3367, especially in the RBD region. Antigenic analysis showed that the S1 domain of LYRa11 could be efficiently recognized by SARS-convalescent human serum, indicating that LYRa11 is a novel virus antigenically close to SARS CoV. Recombination analyses indicate that LYRa11 is likely a recombinant descended from parental lineages that had evolved into a number of bat SARS-like CoVs.
Importance: Although many severe acute respiratory syndrome-like coronaviruses (SARS-like CoVs) have been discovered in bats worldwide, there are significant different genic structures, particularly in the S1 domain, which are responsible for host tropism determination, between bat SARS-like CoVs and human SARS CoVs, indicating that most reported bat SARS-like CoVs are not the progenitors of human SARS CoV. We have identified diverse alphacoronaviruses and a close relative (LYRa11) to SARS CoV in bats collected in Yunnan, China. Further analysis showed that alpha- and betacoronaviruses have different circulation and transmission dynamics in bat populations. Notably, full genomic sequencing and antigenic study demonstrated that LYRa11 is phylogenetically and antigenically closely related to SARS CoV. Recombination analyses indicate that LYRa11 is a recombinant from certain bat SARS-like CoVs circulating in Yunnan Province.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures


Similar articles
-
Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination.J Virol. 2015 Oct;89(20):10532-47. doi: 10.1128/JVI.01048-15. Epub 2015 Aug 12. J Virol. 2015. PMID: 26269185 Free PMC article.
-
Discovery and genetic analysis of novel coronaviruses in least horseshoe bats in southwestern China.Emerg Microbes Infect. 2017 Mar 29;6(3):e14. doi: 10.1038/emi.2016.140. Emerg Microbes Infect. 2017. PMID: 28352124 Free PMC article.
-
Epidemiology and Genomic Characterization of Two Novel SARS-Related Coronaviruses in Horseshoe Bats from Guangdong, China.mBio. 2022 Jun 28;13(3):e0046322. doi: 10.1128/mbio.00463-22. Epub 2022 Apr 25. mBio. 2022. PMID: 35467426 Free PMC article.
-
Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS.Antiviral Res. 2014 Jan;101:45-56. doi: 10.1016/j.antiviral.2013.10.013. Epub 2013 Oct 31. Antiviral Res. 2014. PMID: 24184128 Free PMC article. Review.
-
Bat origin of human coronaviruses.Virol J. 2015 Dec 22;12:221. doi: 10.1186/s12985-015-0422-1. Virol J. 2015. PMID: 26689940 Free PMC article. Review.
Cited by
-
Seroprevalence, cross antigenicity and circulation sphere of bat-borne hantaviruses revealed by serological and antigenic analyses.PLoS Pathog. 2019 Jan 22;15(1):e1007545. doi: 10.1371/journal.ppat.1007545. eCollection 2019 Jan. PLoS Pathog. 2019. PMID: 30668611 Free PMC article.
-
The nervous system during COVID-19: Caught in the crossfire.Immunol Rev. 2022 Oct;311(1):90-111. doi: 10.1111/imr.13114. Epub 2022 Jun 30. Immunol Rev. 2022. PMID: 35770683 Free PMC article. Review.
-
Analysis of the Hosts and Transmission Paths of SARS-CoV-2 in the COVID-19 Outbreak.Genes (Basel). 2020 Jun 9;11(6):637. doi: 10.3390/genes11060637. Genes (Basel). 2020. PMID: 32526937 Free PMC article.
-
Coronaviruses and the human airway: a universal system for virus-host interaction studies.Virol J. 2016 Feb 6;13:24. doi: 10.1186/s12985-016-0479-5. Virol J. 2016. PMID: 26852031 Free PMC article. Review.
-
Using machine learning to detect coronaviruses potentially infectious to humans.Sci Rep. 2023 Jun 8;13(1):9319. doi: 10.1038/s41598-023-35861-7. Sci Rep. 2023. PMID: 37291260 Free PMC article.
References
-
- de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2011. Family Coronaviridae, p 806–828 In King AMQ, Adam MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Academic Press, Ltd., London, United Kingdom
-
- Lai MM, Perlman S, Anderson LJ. 2007. Coronaviridae, p 1304–1335 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, a Wolters Kluwer Business, Philadelphia, PA
-
- Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348:1967–1976. 10.1056/NEJMoa030747 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
