

Regulation of ferroptotic cancer cell death by GPX4
- PMID: 24439385
- PMCID: PMC4076414
- DOI: 10.1016/j.cell.2013.12.010
Regulation of ferroptotic cancer cell death by GPX4
Abstract
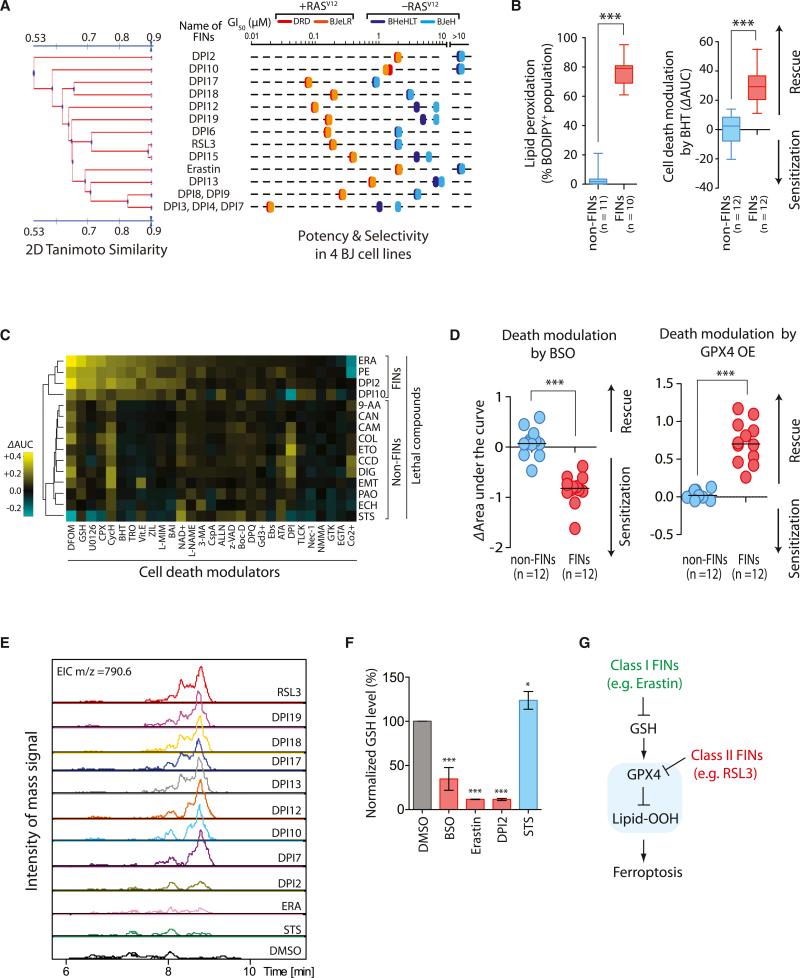
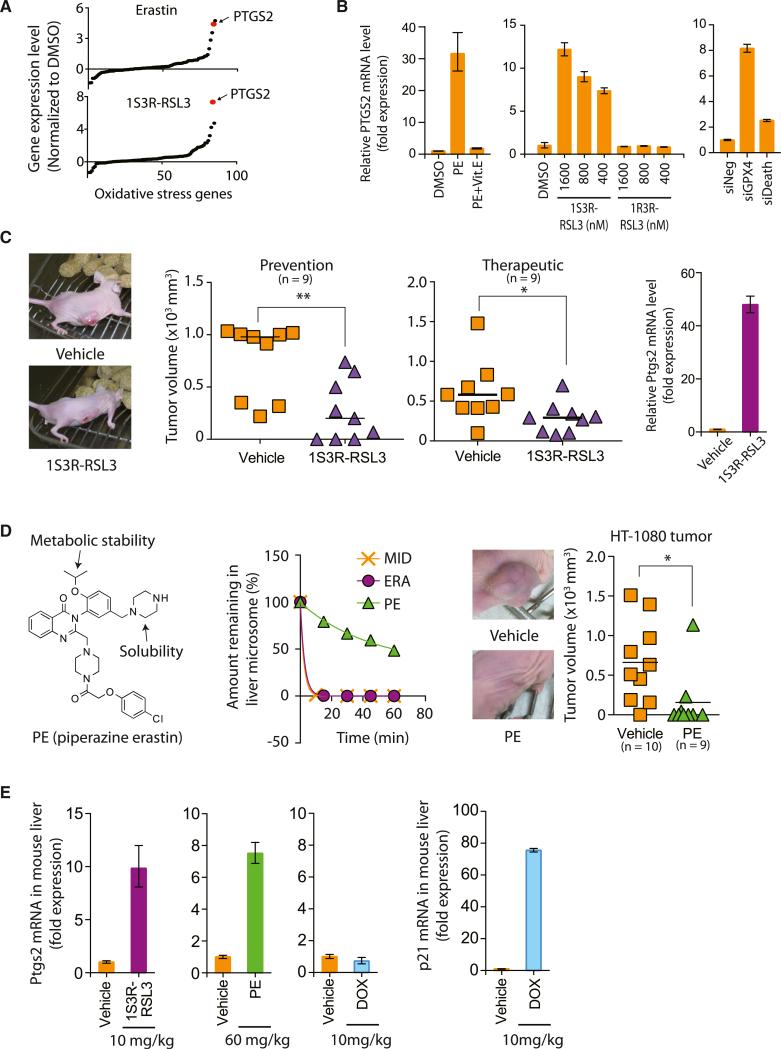
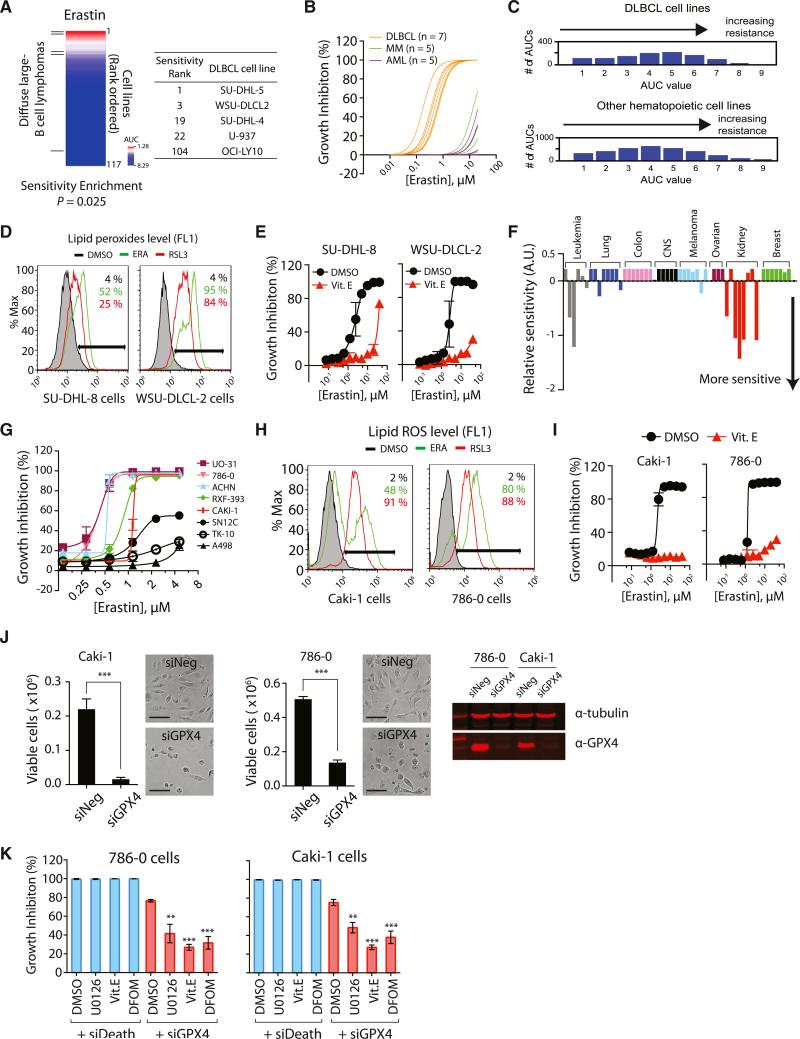
Ferroptosis is a form of nonapoptotic cell death for which key regulators remain unknown. We sought a common mediator for the lethality of 12 ferroptosis-inducing small molecules. We used targeted metabolomic profiling to discover that depletion of glutathione causes inactivation of glutathione peroxidases (GPXs) in response to one class of compounds and a chemoproteomics strategy to discover that GPX4 is directly inhibited by a second class of compounds. GPX4 overexpression and knockdown modulated the lethality of 12 ferroptosis inducers, but not of 11 compounds with other lethal mechanisms. In addition, two representative ferroptosis inducers prevented tumor growth in xenograft mouse tumor models. Sensitivity profiling in 177 cancer cell lines revealed that diffuse large B cell lymphomas and renal cell carcinomas are particularly susceptible to GPX4-regulated ferroptosis. Thus, GPX4 is an essential regulator of ferroptotic cancer cell death.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer.Free Radic Biol Med. 2018 Dec;129:454-462. doi: 10.1016/j.freeradbiomed.2018.10.426. Epub 2018 Oct 16. Free Radic Biol Med. 2018. PMID: 30339884
-
Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis.Curr Top Microbiol Immunol. 2017;403:143-170. doi: 10.1007/82_2016_508. Curr Top Microbiol Immunol. 2017. PMID: 28204974 Review.
-
Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma.Free Radic Biol Med. 2017 Nov;112:597-607. doi: 10.1016/j.freeradbiomed.2017.09.002. Epub 2017 Sep 8. Free Radic Biol Med. 2017. PMID: 28893626 Free PMC article.
-
Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε.FEBS Lett. 2020 Feb;594(4):611-624. doi: 10.1002/1873-3468.13631. Epub 2019 Oct 20. FEBS Lett. 2020. PMID: 31581313
-
Oxytosis/Ferroptosis in Neurodegeneration: the Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4).Mol Neurobiol. 2024 Mar;61(3):1507-1526. doi: 10.1007/s12035-023-03646-8. Epub 2023 Sep 19. Mol Neurobiol. 2024. PMID: 37725216 Review.
Cited by
-
Current progress of ferroptosis study in ovarian cancer.Front Mol Biosci. 2022 Aug 26;9:966007. doi: 10.3389/fmolb.2022.966007. eCollection 2022. Front Mol Biosci. 2022. PMID: 36090052 Free PMC article. Review.
-
Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells.Oncoscience. 2015 May 2;2(5):517-32. doi: 10.18632/oncoscience.160. eCollection 2015. Oncoscience. 2015. PMID: 26097885 Free PMC article.
-
Identification of a group of bisbenzylisoquinoline (BBIQ) compounds as ferroptosis inhibitors.Cell Death Dis. 2022 Nov 26;13(11):1000. doi: 10.1038/s41419-022-05447-8. Cell Death Dis. 2022. PMID: 36435804 Free PMC article.
-
Effects of Natural Products on Enzymes Involved in Ferroptosis: Regulation and Implications.Molecules. 2023 Dec 4;28(23):7929. doi: 10.3390/molecules28237929. Molecules. 2023. PMID: 38067658 Free PMC article. Review.
-
Hesperetin promotes bladder cancer cells death via the PI3K/AKT pathway by network pharmacology and molecular docking.Sci Rep. 2024 Jan 10;14(1):1009. doi: 10.1038/s41598-023-50476-8. Sci Rep. 2024. PMID: 38200039 Free PMC article.
References
-
- Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim. Biophys. Acta. 2013;1830:3289–3303. - PubMed
-
- Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat. Rev. Cancer. 2009;9:501–507. - PubMed
-
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RC2 CA148399/CA/NCI NIH HHS/United States
- 1S10RR025431-01A1/RR/NCRR NIH HHS/United States
- 5R01-CA70823-15/CA/NCI NIH HHS/United States
- S10 RR025431/RR/NCRR NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- 5R01CA097061/CA/NCI NIH HHS/United States
- R01 CA161061/CA/NCI NIH HHS/United States
- R01 GM085081/GM/NIGMS NIH HHS/United States
- RC2-CA148399/CA/NCI NIH HHS/United States
- 5R01GM085081/GM/NIGMS NIH HHS/United States
- R21 CA177591/CA/NCI NIH HHS/United States
- R01 CA097061/CA/NCI NIH HHS/United States
- R01 CA070823/CA/NCI NIH HHS/United States
- R01CA161061/CA/NCI NIH HHS/United States
- ImNIH/Intramural NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
