

Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement
- PMID: 24383718
- PMCID: PMC4543484
- DOI: 10.1089/ars.2013.5814
Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement
Abstract
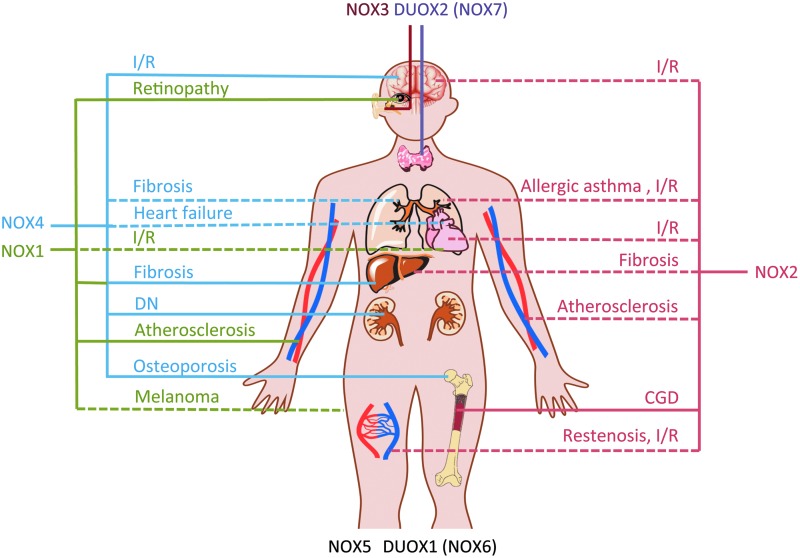
Significance: Oxidative stress, an excess of reactive oxygen species (ROS) production versus consumption, may be involved in the pathogenesis of different diseases. The only known enzymes solely dedicated to ROS generation are nicotinamide adenine dinucleotide phosphate (NADPH) oxidases with their catalytic subunits (NOX). After the clinical failure of most antioxidant trials, NOX inhibitors are the most promising therapeutic option for diseases associated with oxidative stress.
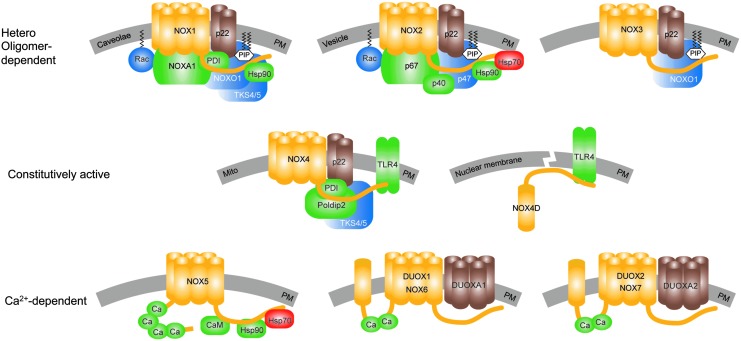
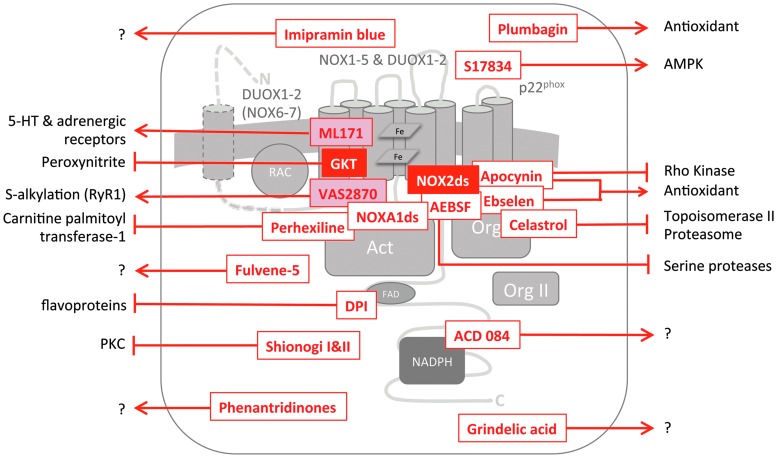
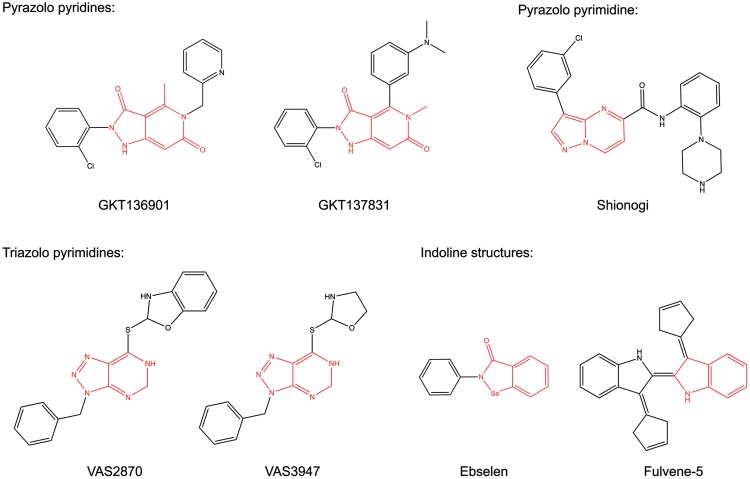
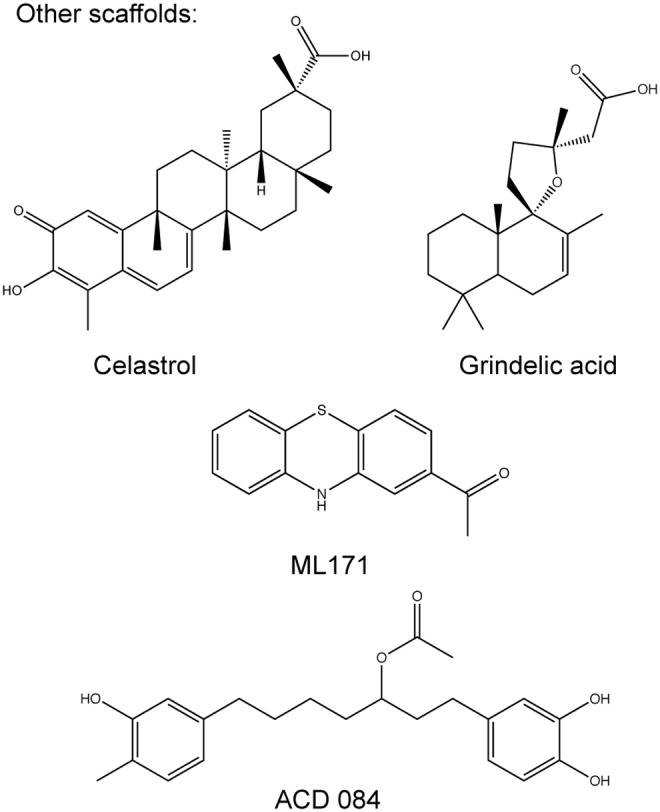
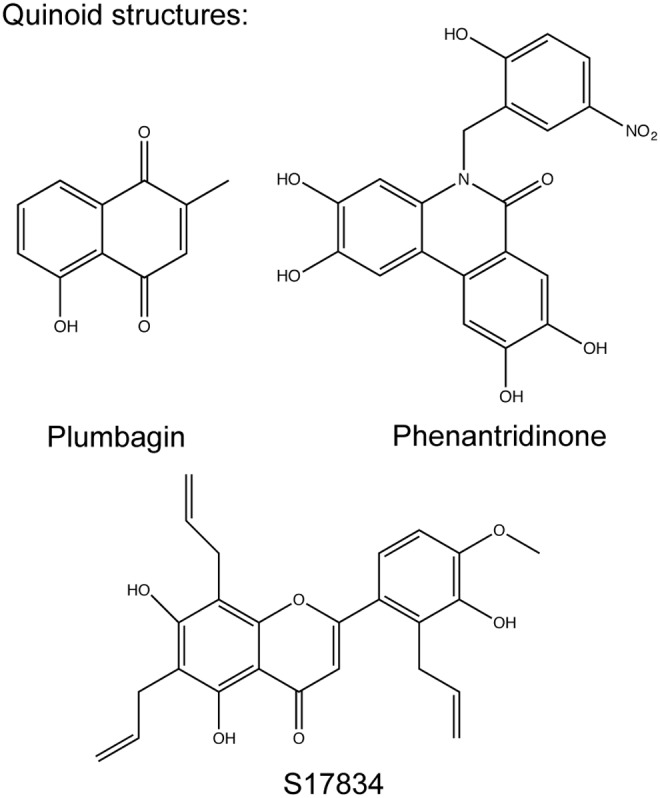
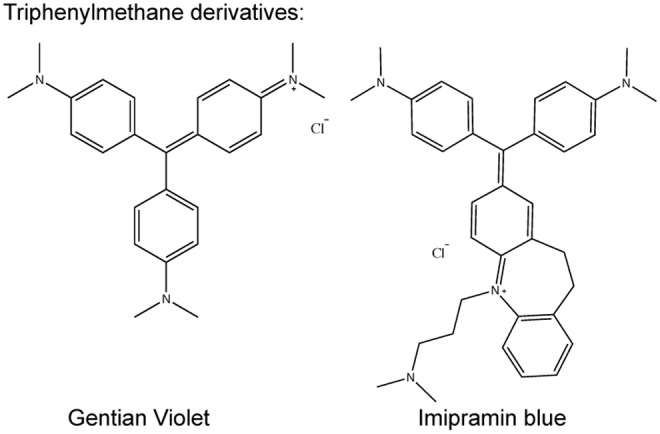
Recent advances: Historical NADPH oxidase inhibitors, apocynin and diphenylene iodonium, are un-specific and not isoform selective. Novel NOX inhibitors stemming from rational drug discovery approaches, for example, GKT137831, ML171, and VAS2870, show improved specificity for NADPH oxidases and moderate NOX isoform selectivity. Along with NOX2 docking sequence (NOX2ds)-tat, a peptide-based inhibitor, the use of these novel small molecules in animal models has provided preliminary in vivo evidence for a pathophysiological role of specific NOX isoforms.
Critical issues: Here, we discuss whether novel NOX inhibitors enable reliable validation of NOX isoforms' pathological roles and whether this knowledge supports translation into pharmacological applications. Modern NOX inhibitors have increased the evidence for pathophysiological roles of NADPH oxidases. However, in comparison to knockout mouse models, NOX inhibitors have limited isoform selectivity. Thus, their use does not enable clear statements on the involvement of individual NOX isoforms in a given disease.
Future directions: The development of isoform-selective NOX inhibitors and biologicals will enable reliable validation of specific NOX isoforms in disease models other than the mouse. Finally, GKT137831, the first NOX inhibitor in clinical development, is poised to provide proof of principle for the clinical potential of NOX inhibition.
Figures
Similar articles
-
NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS.Cell Mol Life Sci. 2012 Jul;69(14):2315-25. doi: 10.1007/s00018-012-1009-2. Epub 2012 May 15. Cell Mol Life Sci. 2012. PMID: 22585059 Free PMC article. Review.
-
Isoform-selective NADPH oxidase inhibitor panel for pharmacological target validation.Free Radic Biol Med. 2020 Feb 20;148:60-69. doi: 10.1016/j.freeradbiomed.2019.12.038. Epub 2019 Dec 25. Free Radic Biol Med. 2020. PMID: 31883469
-
Inhibiting the Activity of NADPH Oxidase in Cancer.Antioxid Redox Signal. 2020 Aug 20;33(6):435-454. doi: 10.1089/ars.2020.8046. Epub 2020 Apr 17. Antioxid Redox Signal. 2020. PMID: 32008376 Free PMC article.
-
The NOX toolbox: validating the role of NADPH oxidases in physiology and disease.Cell Mol Life Sci. 2012 Jul;69(14):2327-43. doi: 10.1007/s00018-012-1010-9. Epub 2012 May 31. Cell Mol Life Sci. 2012. PMID: 22648375 Free PMC article. Review.
-
Pharmacological characterization of the seven human NOX isoforms and their inhibitors.Redox Biol. 2019 Sep;26:101272. doi: 10.1016/j.redox.2019.101272. Epub 2019 Jul 11. Redox Biol. 2019. PMID: 31330481 Free PMC article.
Cited by
-
Redox-based therapeutics in neurodegenerative disease.Br J Pharmacol. 2017 Jun;174(12):1750-1770. doi: 10.1111/bph.13551. Epub 2016 Aug 25. Br J Pharmacol. 2017. PMID: 27477685 Free PMC article. Review.
-
Exploring redox vulnerabilities in JAK2V617F-positive cellular models.Hematol Transfus Cell Ther. 2021 Oct-Dec;43(4):430-436. doi: 10.1016/j.htct.2020.08.006. Epub 2020 Sep 13. Hematol Transfus Cell Ther. 2021. PMID: 32962959 Free PMC article.
-
DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia, and airway remodeling during allergic asthma.JCI Insight. 2016 Nov 3;1(18):e88811. doi: 10.1172/jci.insight.88811. JCI Insight. 2016. PMID: 27812543 Free PMC article.
-
Neutrophils use superoxide to control bacterial infection at a distance.PLoS Pathog. 2018 Jul 17;14(7):e1007157. doi: 10.1371/journal.ppat.1007157. eCollection 2018 Jul. PLoS Pathog. 2018. PMID: 30016370 Free PMC article.
-
Exercise during transition from compensated left ventricular hypertrophy to heart failure in aortic stenosis rats.J Cell Mol Med. 2019 Feb;23(2):1235-1245. doi: 10.1111/jcmm.14025. Epub 2018 Nov 20. J Cell Mol Med. 2019. PMID: 30456799 Free PMC article.
References
-
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, and Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab 9: 686–696, 2008 - PubMed
-
- Anilkumar N, Jose GS, Sawyer I, Santos CXC, Sand C, Brewer AC, Warren D, et al. . A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler Thromb Vasc Biol 33: e104–e112, 2013 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
