

MUC1 in macrophage: contributions to cigarette smoke-induced lung cancer
- PMID: 24282280
- PMCID: PMC3947020
- DOI: 10.1158/0008-5472.CAN-13-1713
MUC1 in macrophage: contributions to cigarette smoke-induced lung cancer
Abstract
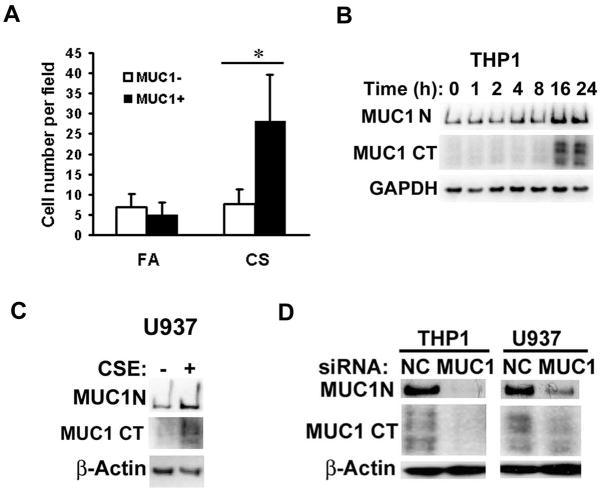
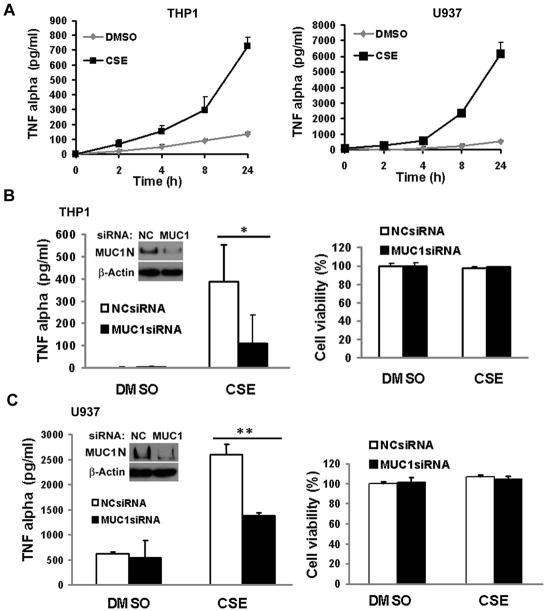
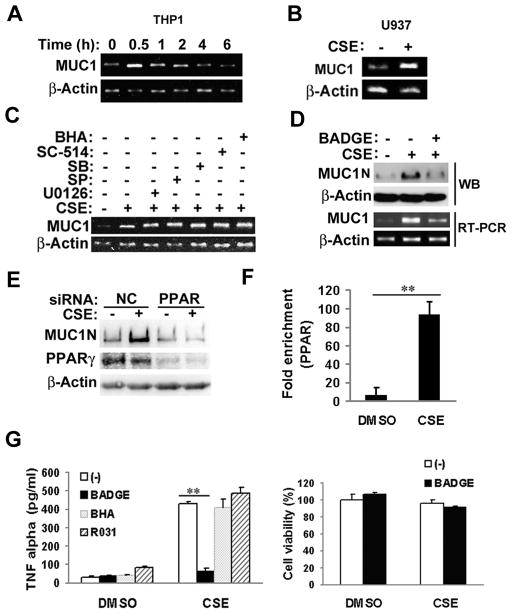
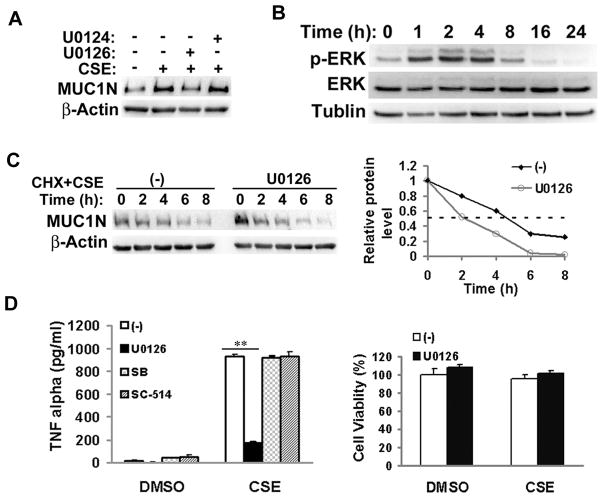
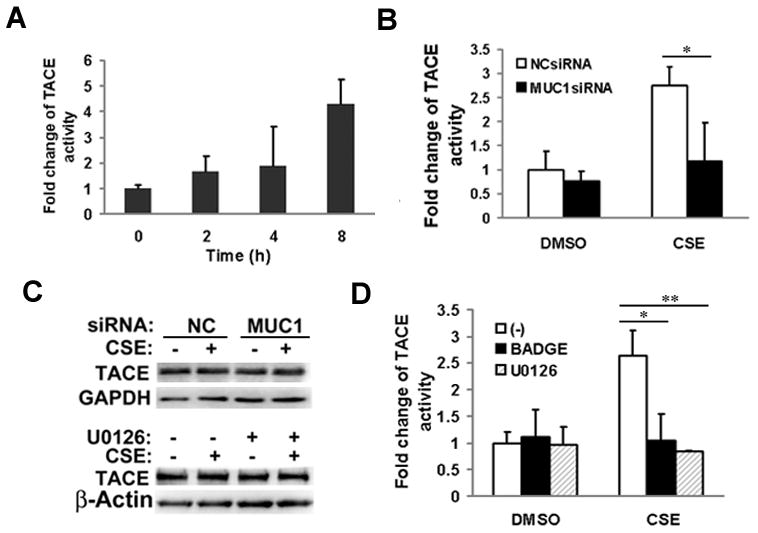
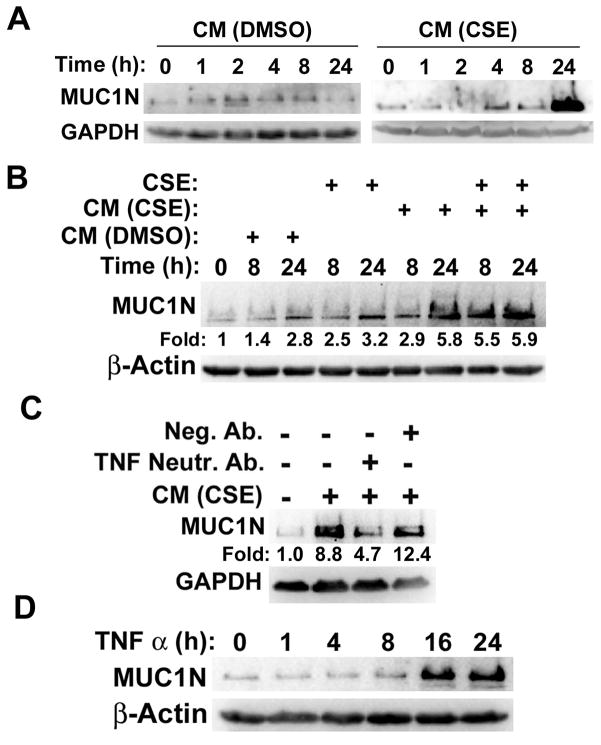
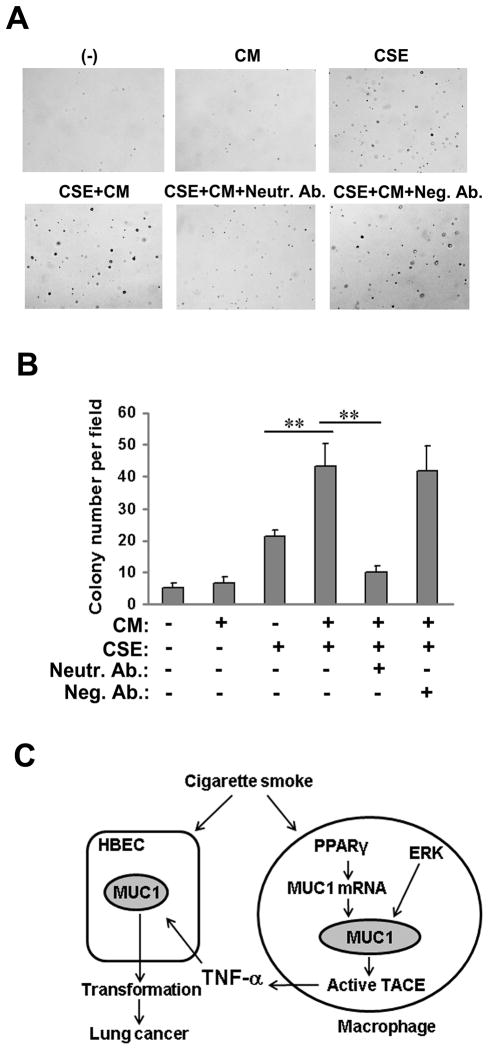
Expression of the pro-oncogenic mucin MUC1 is elevated by inflammation in airway epithelial cells, but the contributions of MUC1 to the development of lung cancer are uncertain. In this study, we developed our finding that cigarette smoke increases Muc1 expression in mouse lung macrophages, where we hypothesized MUC1 may contribute to cigarette smoke-induced transformation of bronchial epithelial cells. In human macrophages, cigarette smoke extract (CSE) strongly induced MUC1 expression through a mechanism involving the nuclear receptor PPAR-γ. CSE-induced extracellular signal-regulated kinase (ERK) activation was also required for MUC1 expression, but it had little effect on MUC1 transcription. RNA interference-mediated attenuation of MUC1 suppressed CSE-induced secretion of TNF-α from macrophages, by suppressing the activity of the TNF-α-converting enzyme (TACE), arguing that MUC1 is required for CSE-induced and TACE-mediated TNF-α secretion. Similarly, MUC1 blockade after CSE induction through suppression of PPAR-γ or ERK inhibited TACE activity and TNF-α secretion. Conditioned media from CSE-treated macrophages induced MUC1 expression and potentiated CSE-induced transformation of human bronchial epithelial cells in a TNF-α-dependent manner. Together, our results identify a signaling pathway involving PPAR-γ, ERK, and MUC1 for TNF-α secretion induced by CSE from macrophages. Furthermore, our results show how MUC1 contributes to smoking-induced lung cancers that are driven by inflammatory signals from macrophages.
Conflict of interest statement
Note: The authors have no conflict of interest to declare.
Figures
Similar articles
-
Up-regulation of TNF-alpha secretion by cigarette smoke is mediated by Egr-1 in HaCaT human keratinocytes.Exp Dermatol. 2010 Aug;19(8):e206-12. doi: 10.1111/j.1600-0625.2009.01050.x. Exp Dermatol. 2010. PMID: 20653771
-
Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract.Toxicol Appl Pharmacol. 2015 Jan 1;282(1):9-19. doi: 10.1016/j.taap.2014.10.022. Epub 2014 Nov 8. Toxicol Appl Pharmacol. 2015. PMID: 25447409
-
Effects of extracellular Hsp70 and cigarette smoke on differentiated THP-1 cells and human monocyte-derived macrophages.Mol Immunol. 2019 Jul;111:53-63. doi: 10.1016/j.molimm.2019.04.002. Epub 2019 Apr 11. Mol Immunol. 2019. PMID: 30981202
-
Muc1 knockout potentiates murine lung carcinogenesis involving an epiregulin-mediated EGFR activation feedback loop.Carcinogenesis. 2017 Jun 1;38(6):604-614. doi: 10.1093/carcin/bgx039. Carcinogenesis. 2017. PMID: 28472347 Free PMC article.
-
Effects of cigarette smoke on Toll-like receptor (TLR) activation of chronic obstructive pulmonary disease (COPD) macrophages.Clin Exp Immunol. 2014 Jun;176(3):461-72. doi: 10.1111/cei.12289. Clin Exp Immunol. 2014. PMID: 24528166 Free PMC article.
Cited by
-
A signaling pathway consisting of miR-551b, catalase and MUC1 contributes to acquired apoptosis resistance and chemoresistance.Carcinogenesis. 2014 Nov;35(11):2457-66. doi: 10.1093/carcin/bgu159. Epub 2014 Aug 1. Carcinogenesis. 2014. PMID: 25085901 Free PMC article.
-
The Role of MRE11 in the IL-6/STAT3 Pathway of Lung Cancer Cells.Curr Issues Mol Biol. 2022 Dec 5;44(12):6132-6144. doi: 10.3390/cimb44120418. Curr Issues Mol Biol. 2022. PMID: 36547079 Free PMC article.
-
Cigarette Smoking-Mediated Macrophage Reprogramming: Mechanistic Insights and Therapeutic Implications.J Nat Sci. 2018 Nov;4(11):e539. J Nat Sci. 2018. PMID: 30801020 Free PMC article.
-
IKBKE Upregulation is Positively Associated with Squamous Cell Carcinoma of the Lung In Vivo and Malignant Transformation of Human Bronchial Epithelial Cells In Vitro.Med Sci Monit. 2015 May 30;21:1577-86. doi: 10.12659/MSM.893815. Med Sci Monit. 2015. PMID: 26025939 Free PMC article.
-
MUC1: The First Respiratory Mucin with an Anti-Inflammatory Function.J Clin Med. 2017 Nov 29;6(12):110. doi: 10.3390/jcm6120110. J Clin Med. 2017. PMID: 29186029 Free PMC article. Review.
References
-
- Cho WC, Kwan CK, Yau S, So PP, Poon PC, Au JS. The role of inflammation in the pathogenesis of lung cancer. Expert opinion on therapeutic targets. 2011;15:1127–37. - PubMed
-
- Milara J, Cortijo J. Tobacco, inflammation, and respiratory tract cancer. Current pharmaceutical design. 2012;18:3901–38. - PubMed
-
- Hecht SS. Tobacco smoke carcinogens and lung cancer. Journal of the National Cancer Institute. 1999;91:1194–210. - PubMed
-
- Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–17. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
