

A role for host activation-induced cytidine deaminase in innate immune defense against KSHV
- PMID: 24244169
- PMCID: PMC3820765
- DOI: 10.1371/journal.ppat.1003748
A role for host activation-induced cytidine deaminase in innate immune defense against KSHV
Abstract
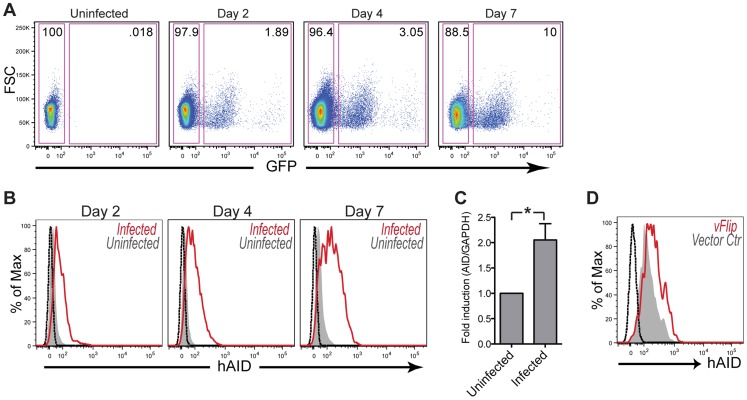
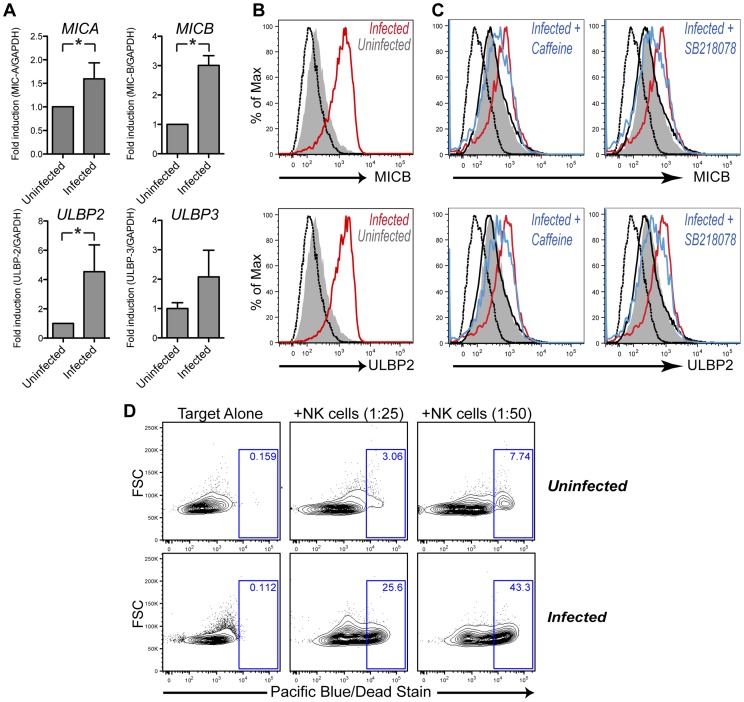
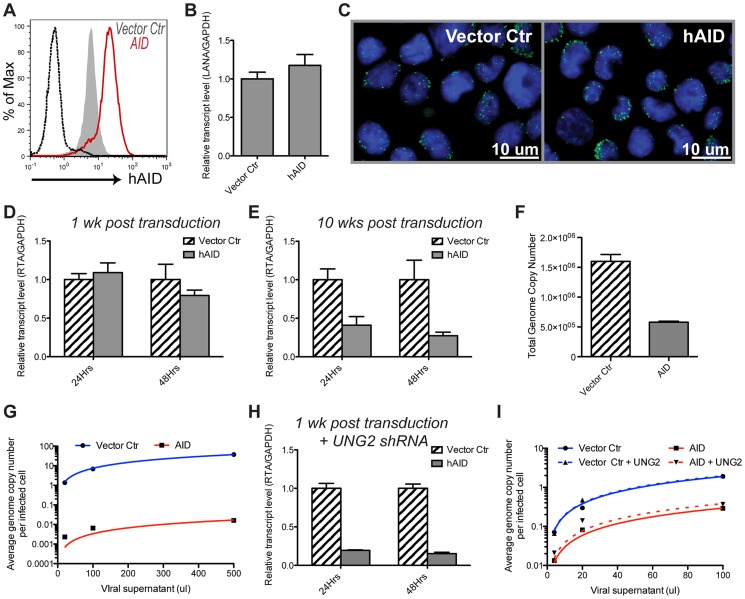
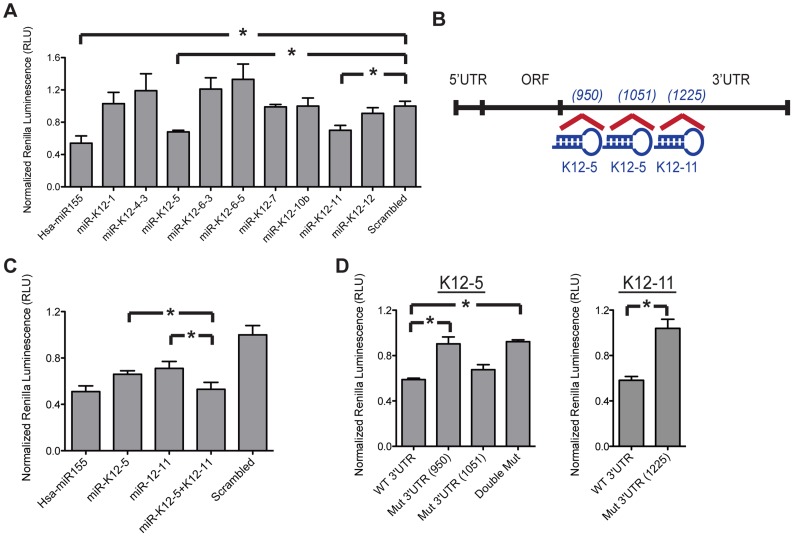
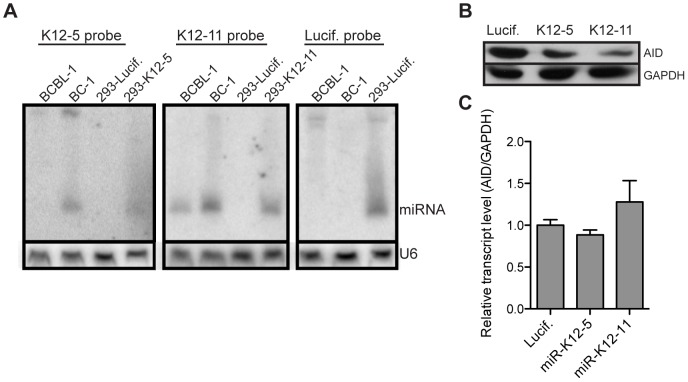
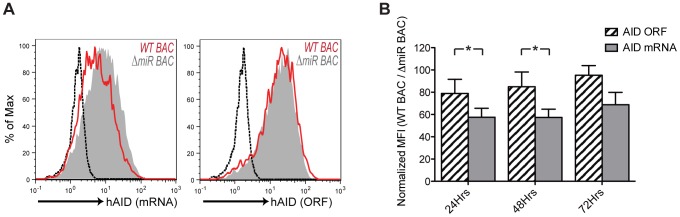
Activation-induced cytidine deaminase (AID) is specifically induced in germinal center B cells to carry out somatic hypermutation and class-switch recombination, two processes responsible for antibody diversification. Because of its mutagenic potential, AID expression and activity are tightly regulated to minimize unwanted DNA damage. Surprisingly, AID expression has been observed ectopically during pathogenic infections. However, the function of AID outside of the germinal centers remains largely uncharacterized. In this study, we demonstrate that infection of human primary naïve B cells with Kaposi's sarcoma-associated herpesvirus (KSHV) rapidly induces AID expression in a cell intrinsic manner. We find that infected cells are marked for elimination by Natural Killer cells through upregulation of NKG2D ligands via the DNA damage pathway, a pathway triggered by AID. Moreover, without having a measurable effect on KSHV latency, AID impinges directly on the viral fitness by inhibiting lytic reactivation and reducing infectivity of KSHV virions. Importantly, we uncover two KSHV-encoded microRNAs that directly regulate AID abundance, further reinforcing the role for AID in the antiviral response. Together our findings reveal additional functions for AID in innate immune defense against KSHV with implications for a broader involvement in innate immunity to other pathogens.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Localization and differential expression of activation-induced cytidine deaminase in the amphibian Xenopus upon antigen stimulation and during early development.J Immunol. 2007 Nov 15;179(10):6783-9. doi: 10.4049/jimmunol.179.10.6783. J Immunol. 2007. PMID: 17982068
-
Killing of Kaposi's sarcoma-associated herpesvirus-infected fibroblasts during latent infection by activated natural killer cells.Eur J Immunol. 2011 Jul;41(7):1958-68. doi: 10.1002/eji.201040661. Epub 2011 May 27. Eur J Immunol. 2011. PMID: 21509779 Free PMC article.
-
Deletion of Murine Gammaherpesvirus Gene M2 in Activation-Induced Cytidine Deaminase-Expressing B Cells Impairs Host Colonization and Viral Reactivation.J Virol. 2020 Dec 9;95(1):e01933-20. doi: 10.1128/JVI.01933-20. Print 2020 Dec 9. J Virol. 2020. PMID: 33028711 Free PMC article.
-
Oncogenic events triggered by AID, the adverse effect of antibody diversification.Carcinogenesis. 2007 Dec;28(12):2427-33. doi: 10.1093/carcin/bgm201. Epub 2007 Sep 4. Carcinogenesis. 2007. PMID: 17804422 Review.
-
Regulation of KSHV Latency and Lytic Reactivation.Viruses. 2020 Sep 17;12(9):1034. doi: 10.3390/v12091034. Viruses. 2020. PMID: 32957532 Free PMC article. Review.
Cited by
-
What's the damage? The impact of pathogens on pathways that maintain host genome integrity.Cell Host Microbe. 2014 Mar 12;15(3):283-94. doi: 10.1016/j.chom.2014.02.010. Cell Host Microbe. 2014. PMID: 24629335 Free PMC article. Review.
-
The Intricate Interplay between APOBEC3 Proteins and DNA Tumour Viruses.Pathogens. 2024 Feb 20;13(3):187. doi: 10.3390/pathogens13030187. Pathogens. 2024. PMID: 38535531 Free PMC article. Review.
-
The Role of APOBECs in Viral Replication.Microorganisms. 2020 Nov 30;8(12):1899. doi: 10.3390/microorganisms8121899. Microorganisms. 2020. PMID: 33266042 Free PMC article. Review.
-
A Protein Antagonist of Activation-Induced Cytidine Deaminase Encoded by a Complex Mouse Retrovirus.mBio. 2019 Aug 13;10(4):e01678-19. doi: 10.1128/mBio.01678-19. mBio. 2019. PMID: 31409681 Free PMC article.
-
Kaposi's Sarcoma-Associated Herpesvirus-Encoded Viral IL-6 (vIL-6) Enhances Immunoglobulin Class-Switch Recombination.Front Microbiol. 2018 Dec 18;9:3119. doi: 10.3389/fmicb.2018.03119. eCollection 2018. Front Microbiol. 2018. PMID: 30619193 Free PMC article.
References
-
- Davison AJ (2002) Evolution of the herpesviruses. Vet Microbiol 86: 69–88. - PubMed
-
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, et al. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266: 1865–1869. - PubMed
-
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, et al. (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86: 1276–1280. - PubMed
-
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332: 1186–1191. - PubMed
-
- Ambroziak JA, Blackbourn DJ, Herndier BG, Glogau RG, Gullett JH, et al. (1995) Herpes-like sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science 268: 582–583. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
