

Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells
- PMID: 23720728
- PMCID: PMC3719826
- DOI: 10.1128/JVI.00805-13
Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells
Erratum in
-
Correction for Ansari et al., "Constitutive Interferon-Inducible Protein 16-Inflammasome Activation during Epstein-Barr Virus Latency I, II, and III in B and Epithelial Cells".J Virol. 2017 Nov 14;91(23):e01519-17. doi: 10.1128/JVI.01519-17. Print 2017 Dec 1. J Virol. 2017. PMID: 29138330 Free PMC article. No abstract available.
Abstract
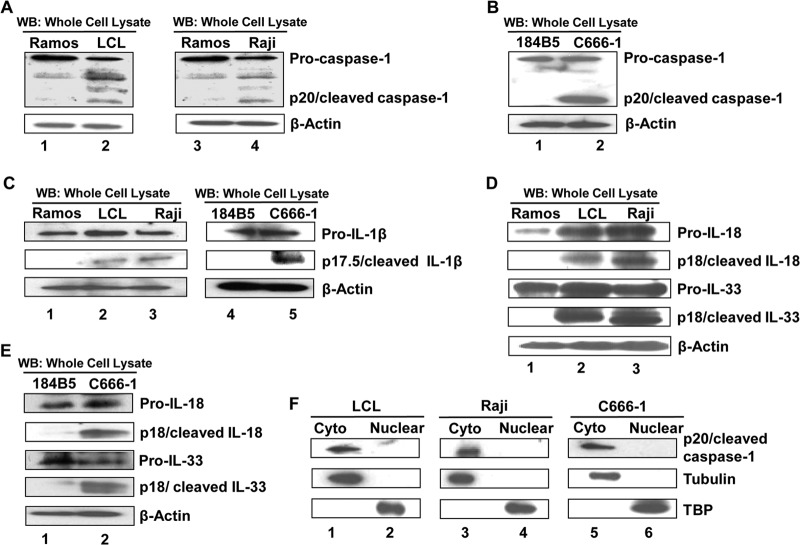
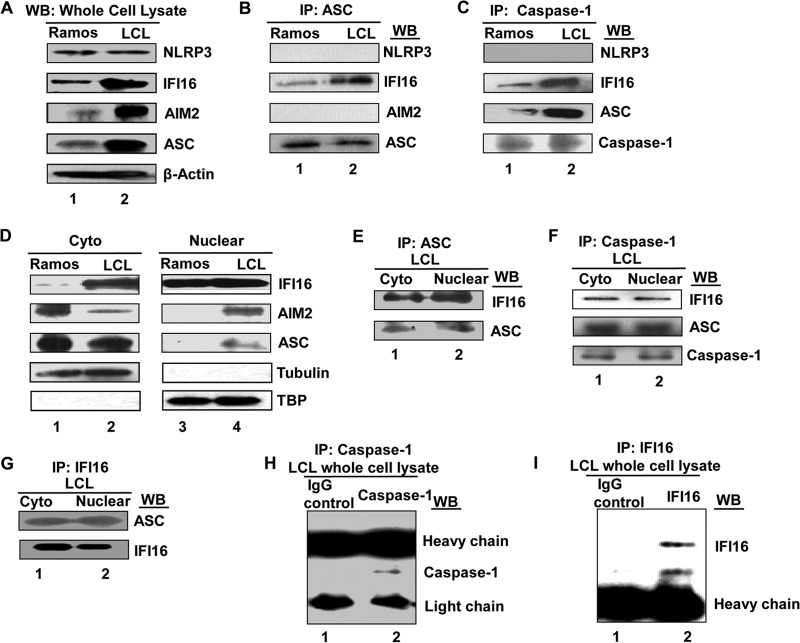
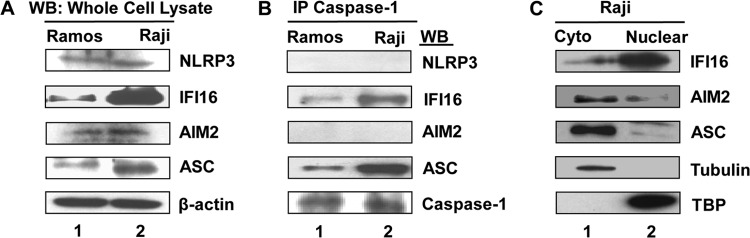
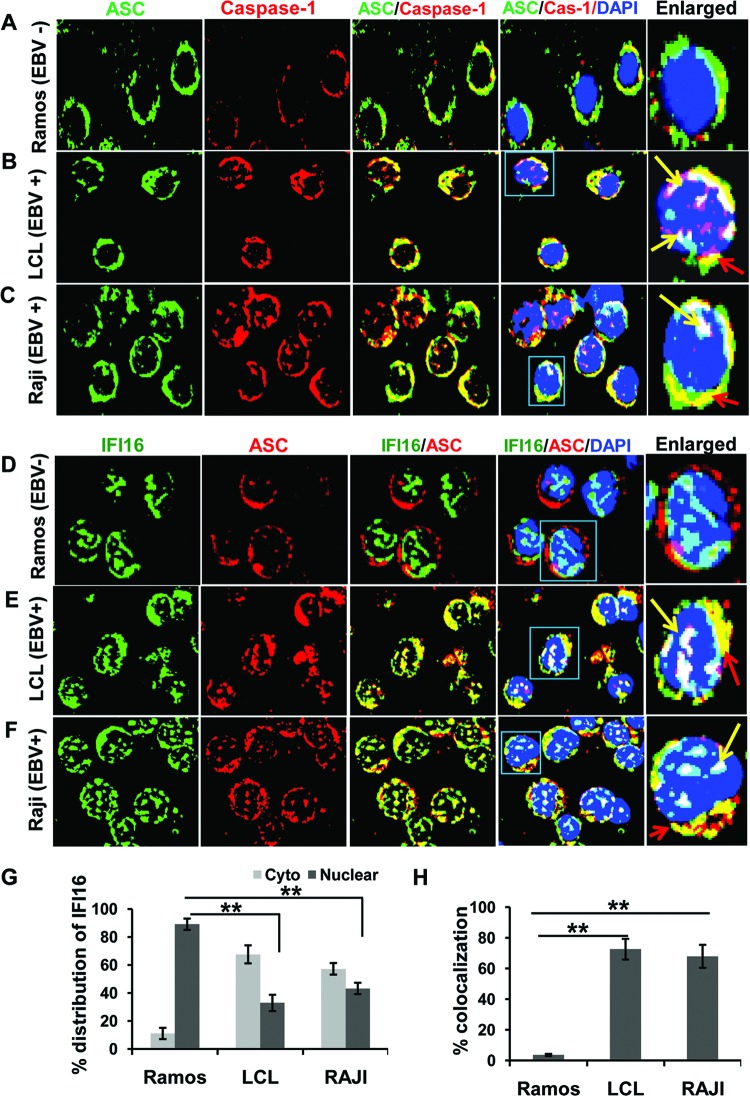
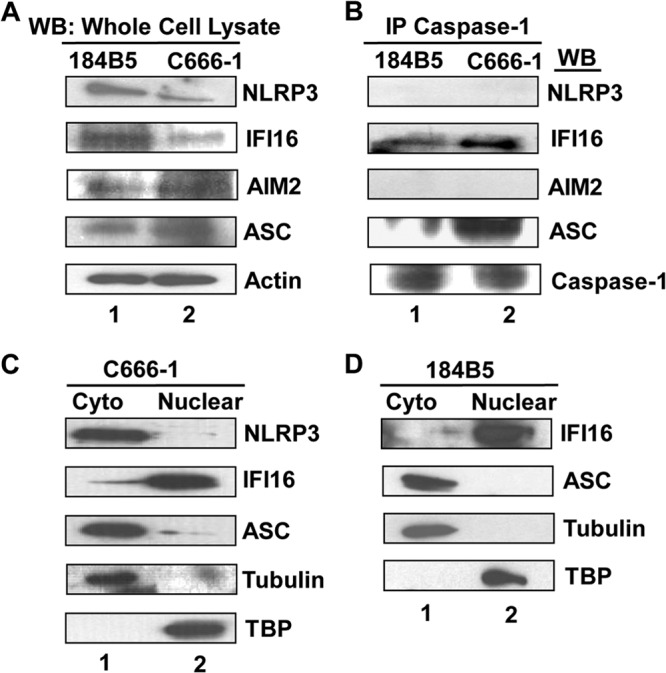
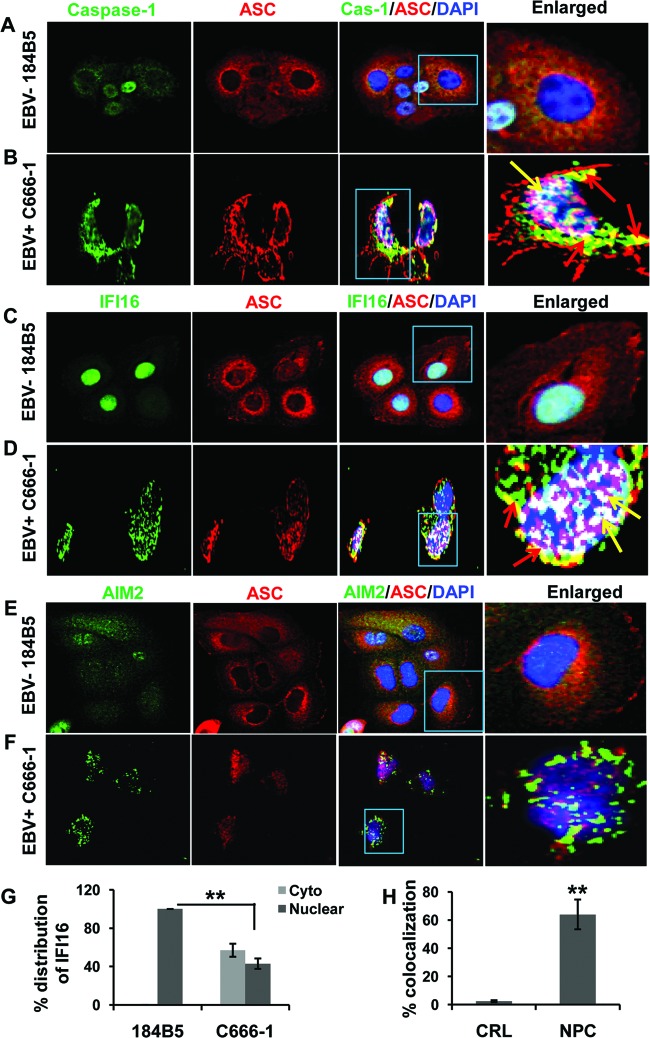
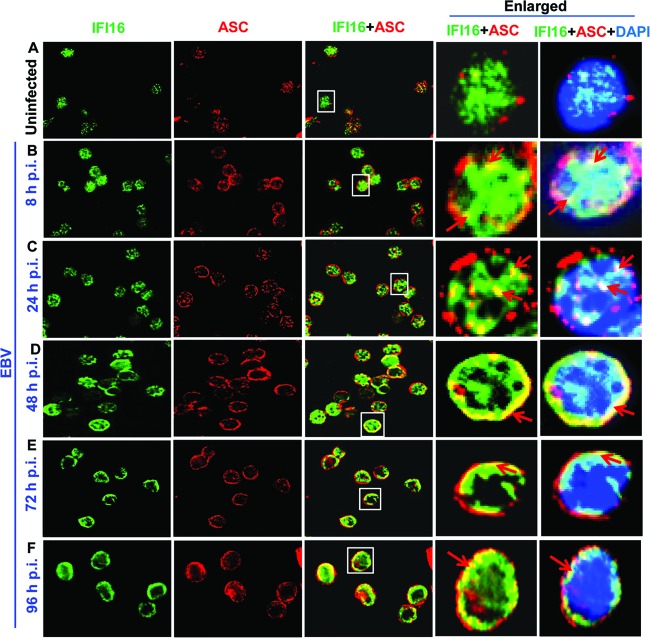
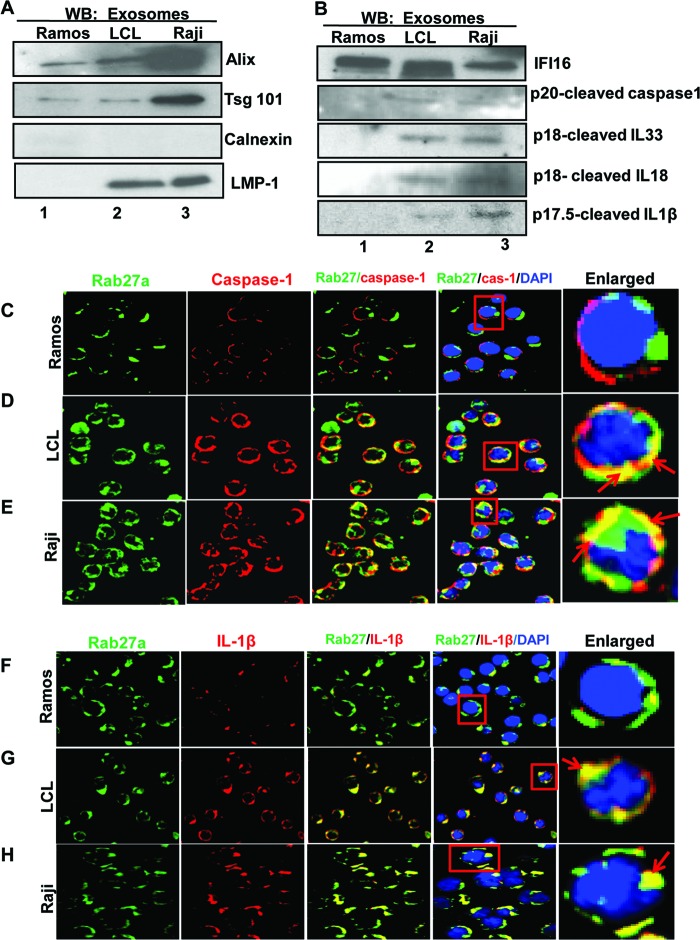
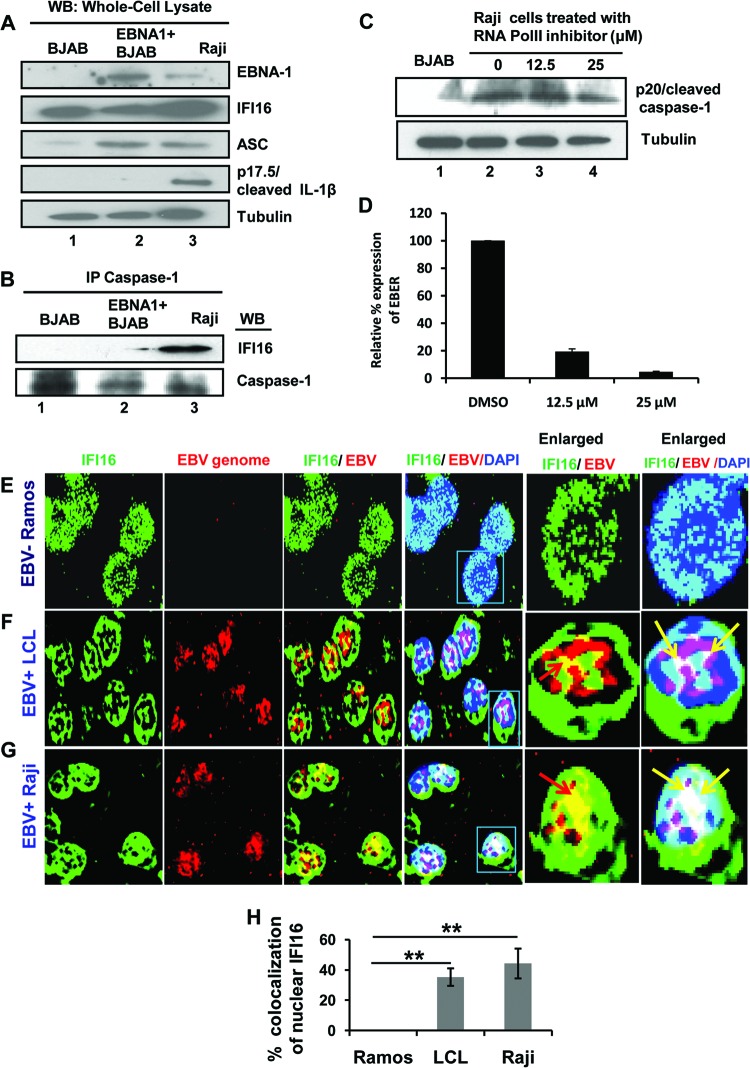
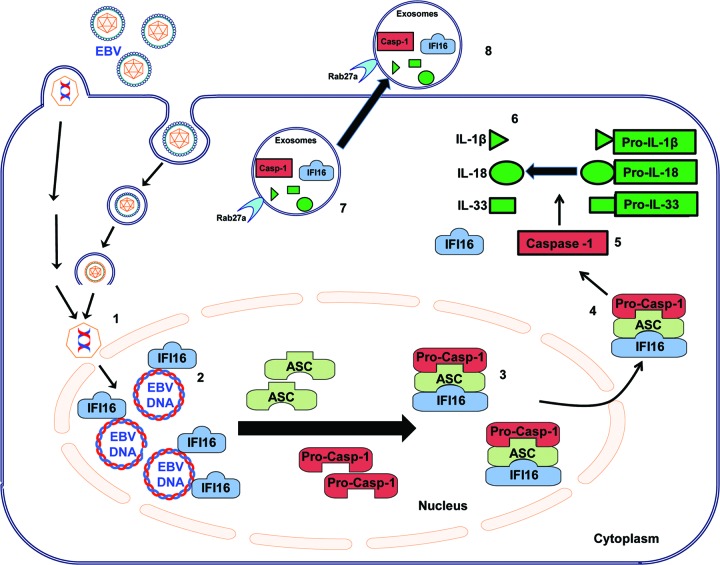
Epstein-Barr virus (EBV), etiologically linked with human B-cell malignancies and nasopharyngeal carcinoma (NPC), establishes three types of latency that facilitate its episomal genome persistence and evasion of host immune responses. The innate inflammasome responses recognize the pathogen-associated molecular patterns which lead into the association of a cytoplasmic sensor such as NLRP3 and AIM2 proteins or nuclear interferon-inducible protein 16 (IFI16) with adaptor ASC protein (apoptosis-associated speck-like protein with a caspase recruitment domain) and effector procaspase-1, resulting in active caspase-1 formation which cleaves the proforms of inflammatory interleukin-1β (IL-1β), IL-18, and IL-33 cytokines. Whether inflammasome responses recognize and respond to EBV genome in the nuclei was not known. We observed evidence of inflammasome activation, such as the activation of caspase-1 and cleavage of pro-IL-1β, -IL-18, and -IL-33, in EBV latency I Raji cells, latency II NPC C666-1 cells, and latency III lymphoblastoid cell lines (LCL). Interaction between ASC with IFI16 but not with AIM2 or NLRP3 was detected in all three latencies and during EBV infection of primary human B cells. IFI16 and cleaved caspase-1, IL-1β, IL-18, and IL-33 were detected in the exosomes from Raji cells and LCL. Though EBV nuclear antigen 1 (EBNA1) and EBV-encoded small RNAs (EBERs) are common to all forms of EBV latency, caspase-1 cleavage was not detected in cells expressing EBNA1 alone, and blocking EBER transcription did not inhibit caspase-1 cleavage. In fluorescence in situ hybridization (FISH) analysis, IFI16 colocalized with the EBV genome in LCL and Raji cell nuclei. These studies demonstrated that constant sensing of latent EBV genome by IFI16 in all types of latency results in the constitutive induction of the inflammasome and IL-1β, IL-18, and IL-33 maturation.
Figures
Similar articles
-
Kaposi's sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes.J Virol. 2013 Apr;87(8):4417-31. doi: 10.1128/JVI.03282-12. Epub 2013 Feb 6. J Virol. 2013. PMID: 23388709 Free PMC article.
-
Epstein-Barr virus infection-induced inflammasome activation in human monocytes.PLoS One. 2017 Apr 3;12(4):e0175053. doi: 10.1371/journal.pone.0175053. eCollection 2017. PLoS One. 2017. PMID: 28369146 Free PMC article.
-
BRCA1 Regulates IFI16 Mediated Nuclear Innate Sensing of Herpes Viral DNA and Subsequent Induction of the Innate Inflammasome and Interferon-β Responses.PLoS Pathog. 2015 Jun 29;11(6):e1005030. doi: 10.1371/journal.ppat.1005030. eCollection 2015 Jun. PLoS Pathog. 2015. Retraction in: PLoS Pathog. 2022 Oct 19;18(10):e1010904. doi: 10.1371/journal.ppat.1010904. PMID: 26121674 Free PMC article. Retracted.
-
Epstein-Barr virus latency: LMP2, a regulator or means for Epstein-Barr virus persistence?Adv Cancer Res. 2000;79:175-200. doi: 10.1016/s0065-230x(00)79006-3. Adv Cancer Res. 2000. PMID: 10818681 Review.
-
Regulation and dysregulation of Epstein-Barr virus latency: implications for the development of autoimmune diseases.Autoimmunity. 2008 May;41(4):298-328. doi: 10.1080/08916930802024772. Autoimmunity. 2008. PMID: 18432410 Review.
Cited by
-
Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape.World J Virol. 2016 Aug 12;5(3):87-96. doi: 10.5501/wjv.v5.i3.87. World J Virol. 2016. PMID: 27563536 Free PMC article. Review.
-
T-Lymphocyte Activation Is Correlated With the Presence of Anti-EBV in Patients With Laryngeal Squamous Cell Carcinoma.In Vivo. 2019 Nov-Dec;33(6):2007-2012. doi: 10.21873/invivo.11697. In Vivo. 2019. PMID: 31662531 Free PMC article.
-
Inflammation and its role in age-related macular degeneration.Cell Mol Life Sci. 2016 May;73(9):1765-86. doi: 10.1007/s00018-016-2147-8. Epub 2016 Feb 6. Cell Mol Life Sci. 2016. PMID: 26852158 Free PMC article. Review.
-
Exosome Biogenesis, Regulation, and Function in Viral Infection.Viruses. 2015 Sep 17;7(9):5066-83. doi: 10.3390/v7092862. Viruses. 2015. PMID: 26393640 Free PMC article. Review.
-
Interplay between hypoxia and inflammation contributes to the progression and severity of respiratory viral diseases.Mol Aspects Med. 2021 Oct;81:101000. doi: 10.1016/j.mam.2021.101000. Epub 2021 Jul 19. Mol Aspects Med. 2021. PMID: 34294412 Free PMC article. Review.
References
-
- Maeda E, Akahane M, Kiryu S, Kato N, Yoshikawa T, Hayashi N, Aoki S, Minami M, Uozaki H, Fukayama M, Ohtomo K. 2009. Spectrum of Epstein-Barr virus-related diseases: a pictorial review. Jpn. J. Radiol. 27:4–19 - PubMed
-
- Amon W, Farrell PJ. 2005. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 15:149–156 - PubMed
-
- Young LS, Arrand JR, Murray PG. 2007. EBV gene expression and regulation, p 461–489 Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
