

The pathogenesis of cardiac fibrosis
- PMID: 23649149
- PMCID: PMC3769482
- DOI: 10.1007/s00018-013-1349-6
The pathogenesis of cardiac fibrosis
Abstract
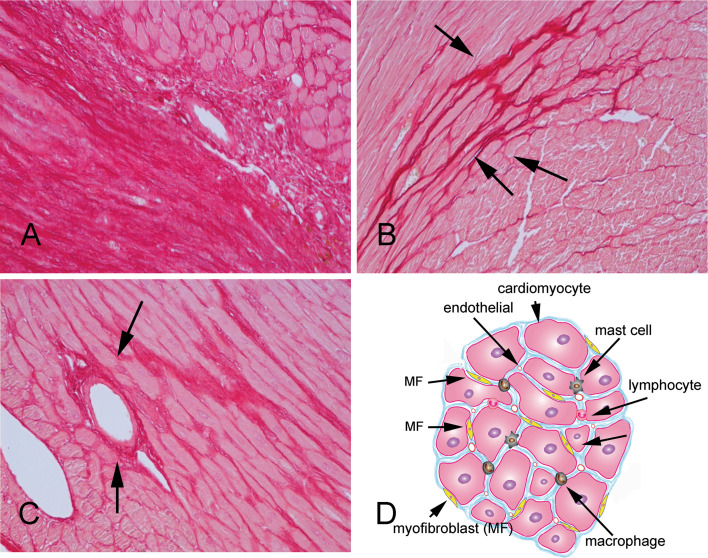
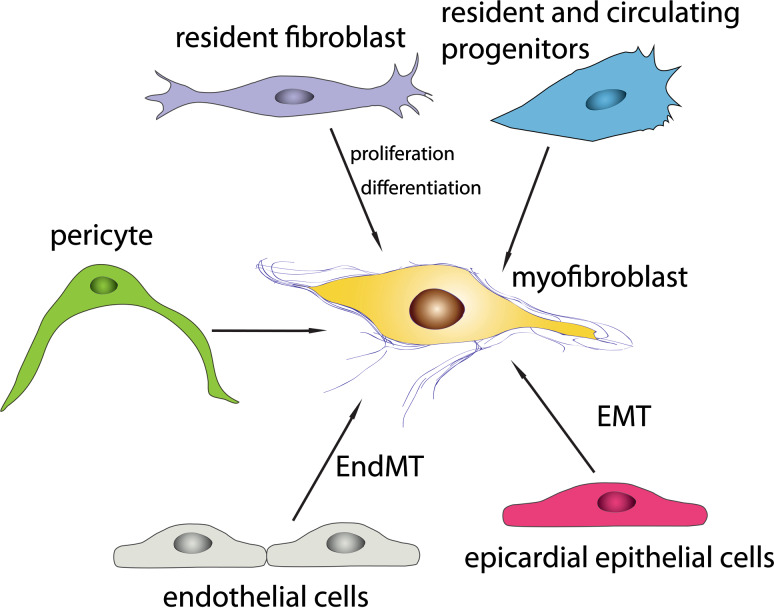
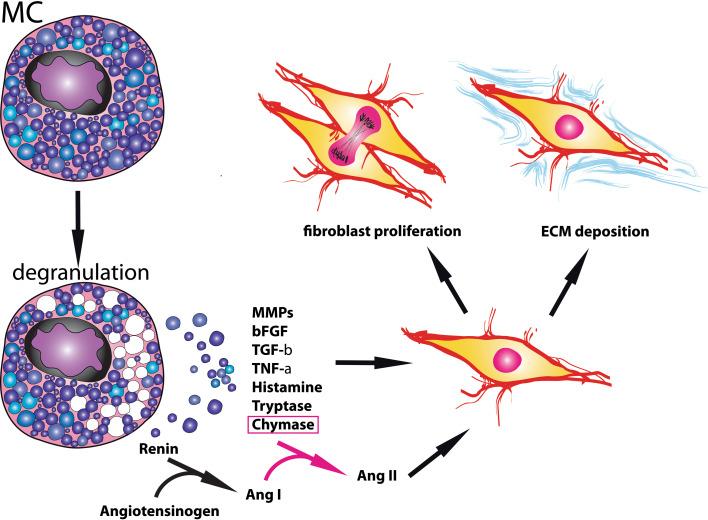
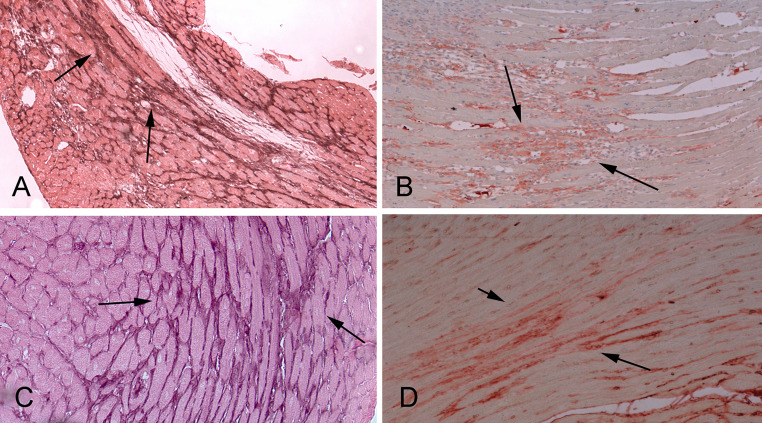
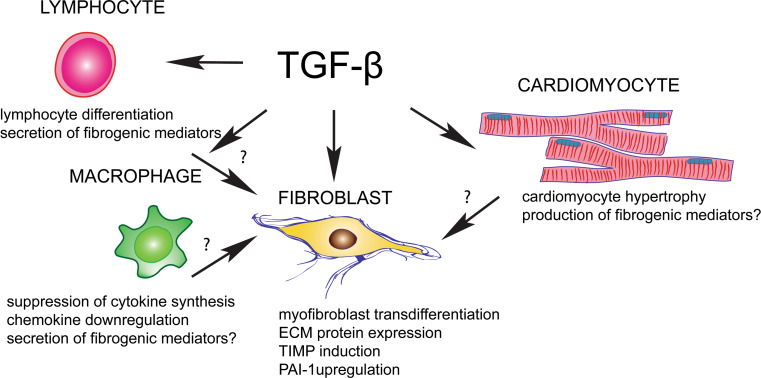
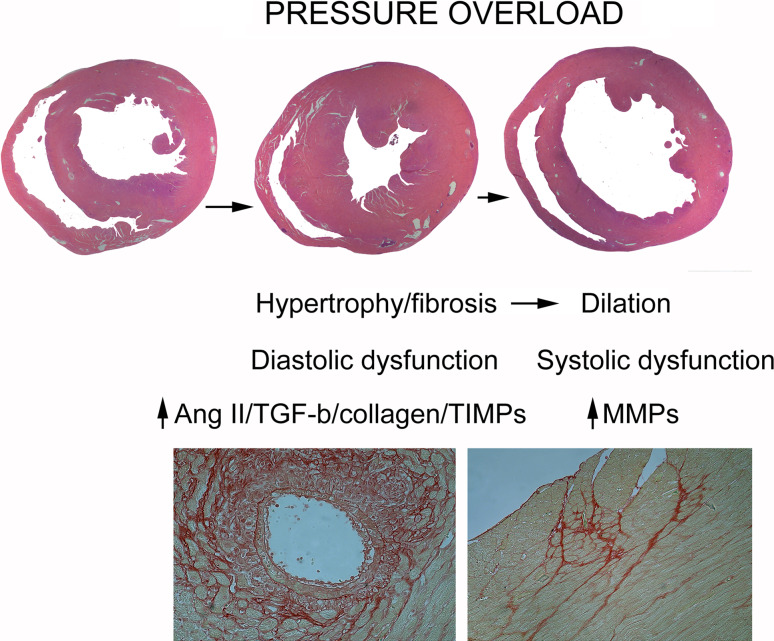
Cardiac fibrosis is characterized by net accumulation of extracellular matrix proteins in the cardiac interstitium, and contributes to both systolic and diastolic dysfunction in many cardiac pathophysiologic conditions. This review discusses the cellular effectors and molecular pathways implicated in the pathogenesis of cardiac fibrosis. Although activated myofibroblasts are the main effector cells in the fibrotic heart, monocytes/macrophages, lymphocytes, mast cells, vascular cells and cardiomyocytes may also contribute to the fibrotic response by secreting key fibrogenic mediators. Inflammatory cytokines and chemokines, reactive oxygen species, mast cell-derived proteases, endothelin-1, the renin/angiotensin/aldosterone system, matricellular proteins, and growth factors (such as TGF-β and PDGF) are some of the best-studied mediators implicated in cardiac fibrosis. Both experimental and clinical evidence suggests that cardiac fibrotic alterations may be reversible. Understanding the mechanisms responsible for initiation, progression, and resolution of cardiac fibrosis is crucial to design anti-fibrotic treatment strategies for patients with heart disease.
Figures
Similar articles
-
Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities.Mol Aspects Med. 2019 Feb;65:70-99. doi: 10.1016/j.mam.2018.07.001. Epub 2018 Aug 2. Mol Aspects Med. 2019. PMID: 30056242 Review.
-
Innate Immunity Effector Cells as Inflammatory Drivers of Cardiac Fibrosis.Int J Mol Sci. 2020 Sep 28;21(19):7165. doi: 10.3390/ijms21197165. Int J Mol Sci. 2020. PMID: 32998408 Free PMC article. Review.
-
Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities.J Mol Cell Cardiol. 2016 Jan;90:84-93. doi: 10.1016/j.yjmcc.2015.12.011. Epub 2015 Dec 15. J Mol Cell Cardiol. 2016. PMID: 26705059 Free PMC article. Review.
-
Fibroblasts and the extracellular matrix in right ventricular disease.Cardiovasc Res. 2017 Oct 1;113(12):1453-1464. doi: 10.1093/cvr/cvx146. Cardiovasc Res. 2017. PMID: 28957531 Free PMC article. Review.
-
Plasminogen activator inhibitor-1 reduces cardiac fibrosis and promotes M2 macrophage polarization in inflammatory cardiomyopathy.Basic Res Cardiol. 2021 Jan 11;116(1):1. doi: 10.1007/s00395-020-00840-w. Basic Res Cardiol. 2021. PMID: 33432417 Free PMC article.
Cited by
-
Assessment of Myocardial Diastolic Dysfunction as a Result of Myocardial Infarction and Extracellular Matrix Regulation Disorders in the Context of Mesenchymal Stem Cell Therapy.J Clin Med. 2022 Sep 15;11(18):5430. doi: 10.3390/jcm11185430. J Clin Med. 2022. PMID: 36143077 Free PMC article. Review.
-
lncRNA HOTAIR and Cardiovascular diseases.Funct Integr Genomics. 2024 Sep 19;24(5):165. doi: 10.1007/s10142-024-01444-6. Funct Integr Genomics. 2024. PMID: 39294422 Review.
-
Antihypertensive and Antifibrosis Effects of Acupuncture at PC6 Acupoints in Spontaneously Hypertensive Rats and the Underlying Mechanisms.Front Physiol. 2020 Aug 26;11:734. doi: 10.3389/fphys.2020.00734. eCollection 2020. Front Physiol. 2020. PMID: 32982761 Free PMC article.
-
Fibrous Remodeling in Eosinophilic Esophagitis: Clinical Facts and Pathophysiological Uncertainties.Int J Mol Sci. 2024 Jan 11;25(2):927. doi: 10.3390/ijms25020927. Int J Mol Sci. 2024. PMID: 38256003 Free PMC article. Review.
-
SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation.Basic Res Cardiol. 2021 Apr 13;116(1):25. doi: 10.1007/s00395-021-00865-9. Basic Res Cardiol. 2021. PMID: 33847835 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
