

Epigenetic diversity of Kaposi's sarcoma-associated herpesvirus
- PMID: 23361465
- PMCID: PMC3597696
- DOI: 10.1093/nar/gkt033
Epigenetic diversity of Kaposi's sarcoma-associated herpesvirus
Abstract
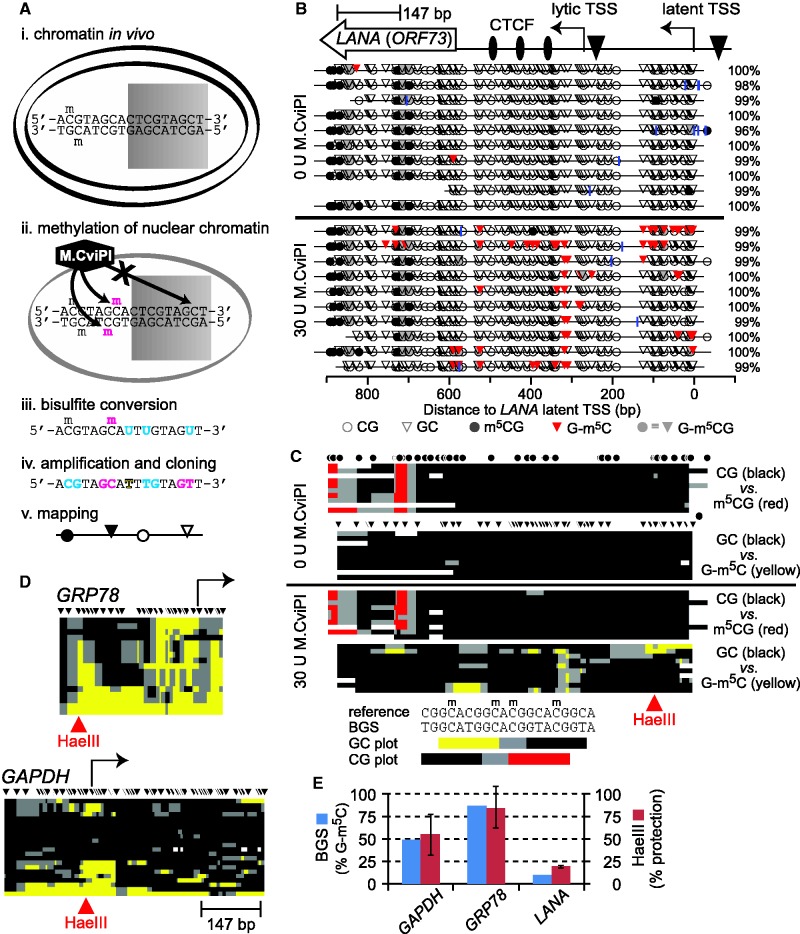
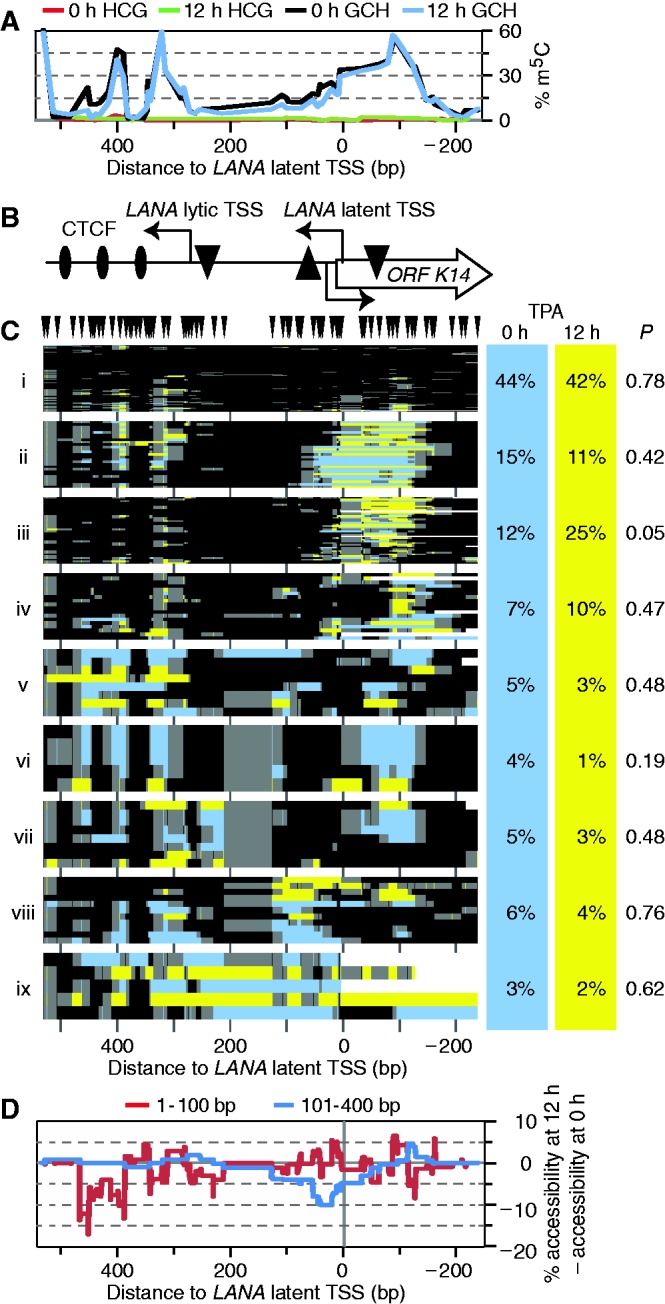
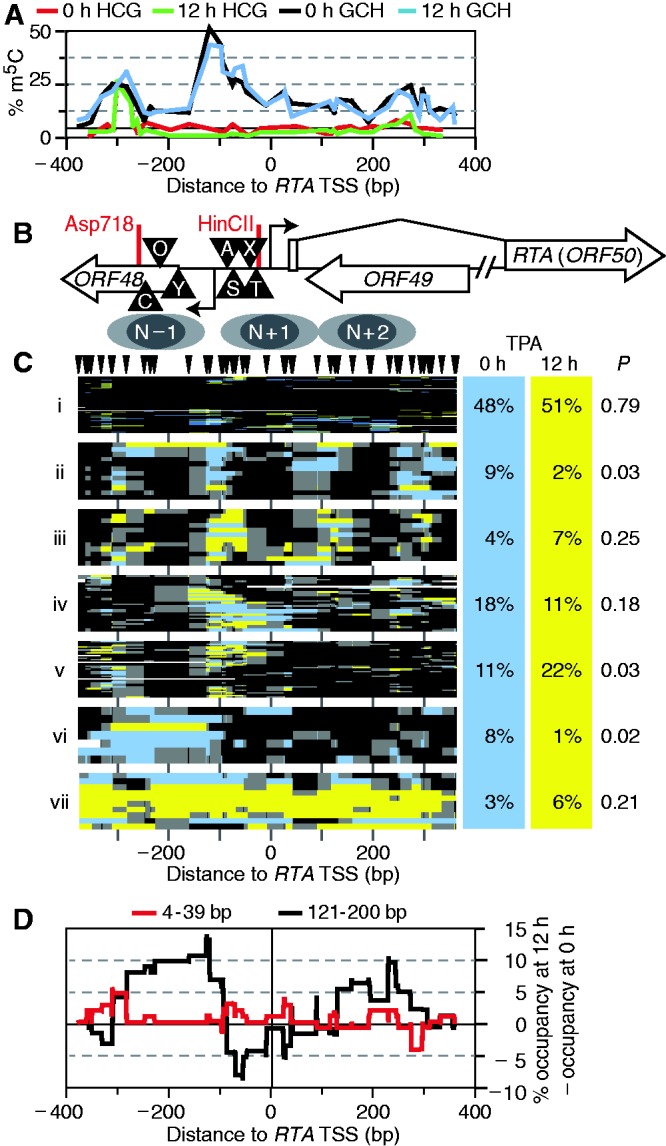
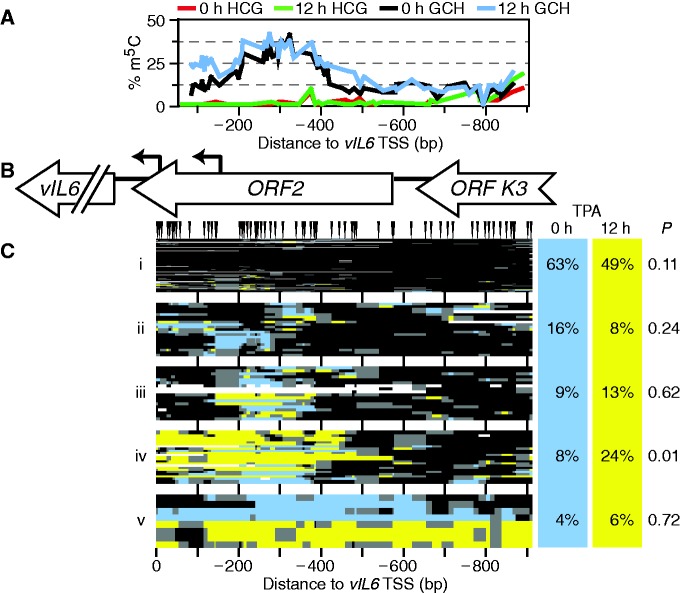
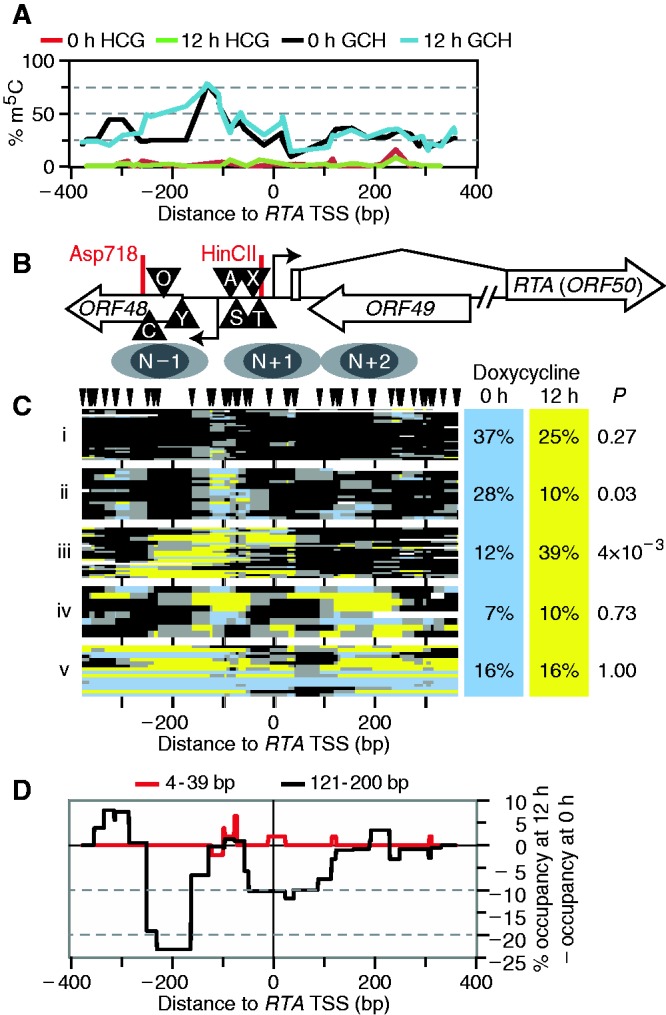
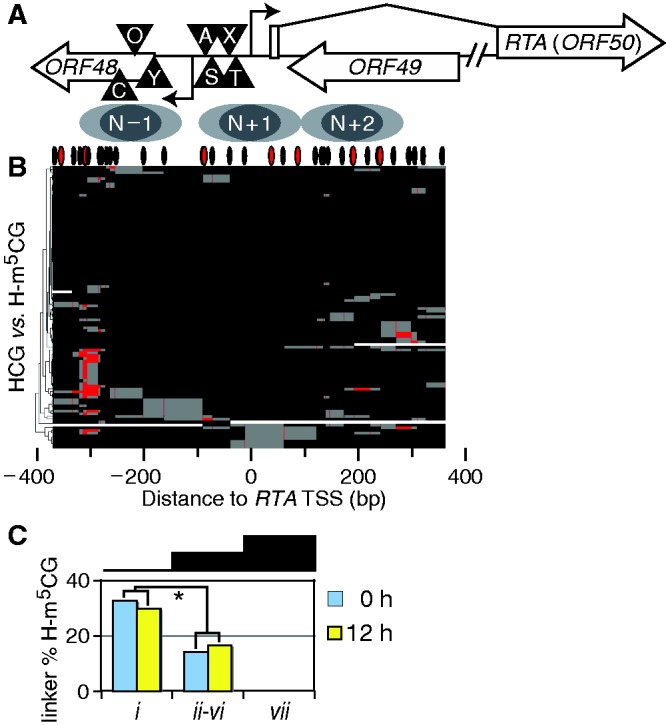
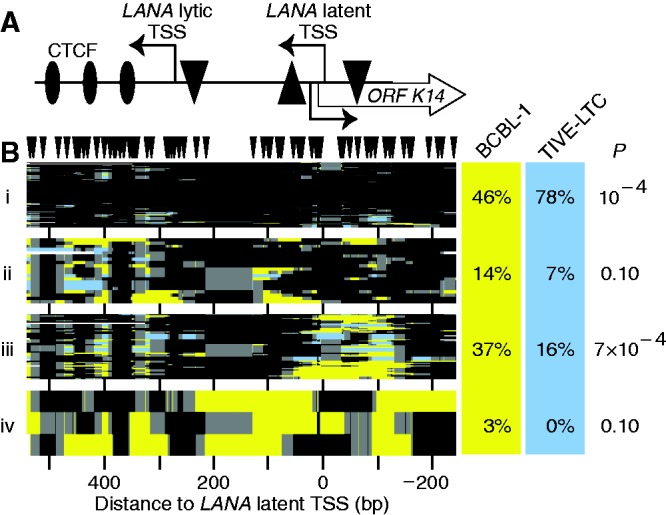
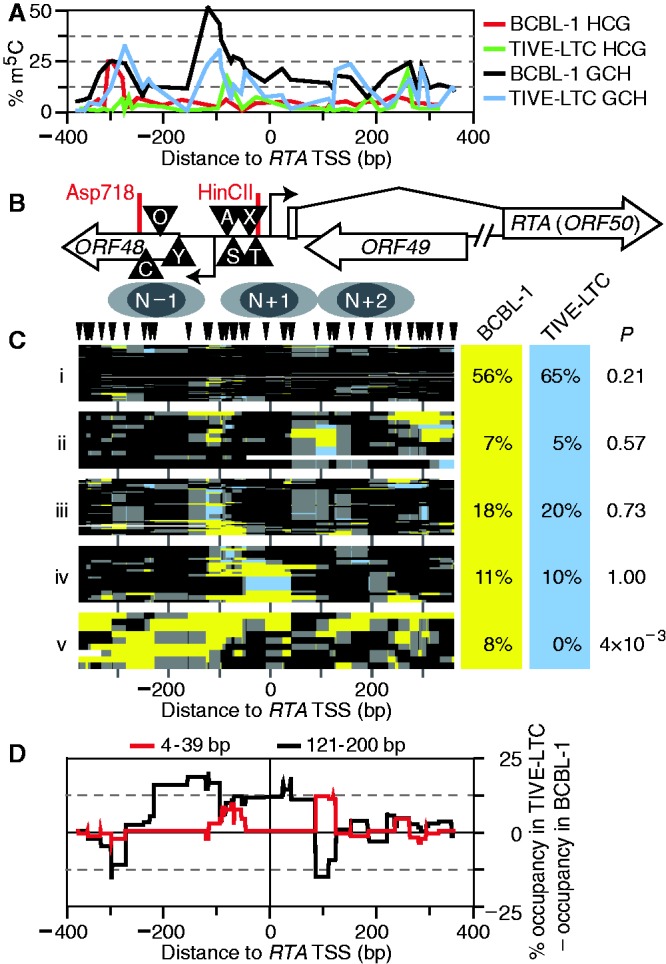
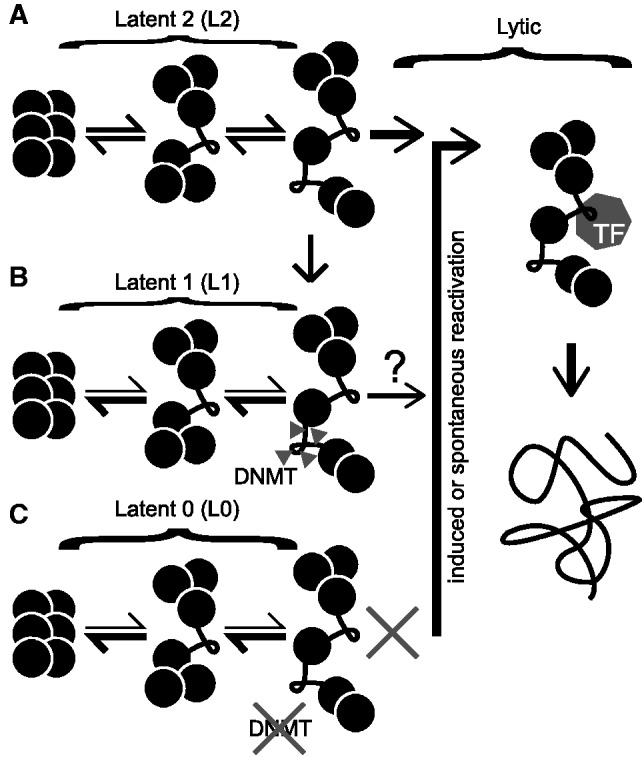
Spontaneous lytic reactivation of Kaposi's sarcoma-associated herpesvirus (KSHV) occurs at a low rate in latently infected cells in disease and culture. This suggests imperfect epigenetic maintenance of viral transcription programs, perhaps due to variability in chromatin structure at specific loci across the population of KSHV episomal genomes. To characterize this locus-specific chromatin structural diversity, we used MAPit single-molecule footprinting, which simultaneously maps endogenous CG methylation and accessibility to M.CviPI at GC sites. Diverse chromatin structures were detected at the LANA, RTA and vIL6 promoters. At each locus, chromatin ranged from fully closed to fully open across the population. This diversity has not previously been reported in a virus. Phorbol ester and RTA transgene induction were used to identify chromatin conformations associated with reactivation of lytic transcription, which only a fraction of episomes had. Moreover, certain chromatin conformations correlated with CG methylation patterns at the RTA and vIL6 promoters. This indicated that some of the diverse chromatin conformations at these loci were epigenetically distinct. Finally, by comparing chromatin structures from a cell line infected with constitutively latent virus, we identified products of lytic replication. Our findings show that epigenetic drift can restrict viral propagation by chromatin compaction at latent and lytic promoters.
Figures
Similar articles
-
Chromatin remodeling controls Kaposi's sarcoma-associated herpesvirus reactivation from latency.PLoS Pathog. 2018 Sep 13;14(9):e1007267. doi: 10.1371/journal.ppat.1007267. eCollection 2018 Sep. PLoS Pathog. 2018. PMID: 30212584 Free PMC article.
-
BET-Inhibitors Disrupt Rad21-Dependent Conformational Control of KSHV Latency.PLoS Pathog. 2017 Jan 20;13(1):e1006100. doi: 10.1371/journal.ppat.1006100. eCollection 2017 Jan. PLoS Pathog. 2017. PMID: 28107481 Free PMC article.
-
Genome-Wide Identification of Direct RTA Targets Reveals Key Host Factors for Kaposi's Sarcoma-Associated Herpesvirus Lytic Reactivation.J Virol. 2019 Feb 19;93(5):e01978-18. doi: 10.1128/JVI.01978-18. Print 2019 Mar 1. J Virol. 2019. PMID: 30541837 Free PMC article.
-
Molecular biology of KSHV lytic reactivation.Viruses. 2015 Jan 14;7(1):116-53. doi: 10.3390/v7010116. Viruses. 2015. PMID: 25594835 Free PMC article. Review.
-
Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus replication.Semin Cancer Biol. 2009 Jun;19(3):153-7. doi: 10.1016/j.semcancer.2009.02.010. Epub 2009 Feb 21. Semin Cancer Biol. 2009. PMID: 19429478 Free PMC article. Review.
Cited by
-
LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex.PLoS Pathog. 2014 Jul 17;10(7):e1004240. doi: 10.1371/journal.ppat.1004240. eCollection 2014 Jul. PLoS Pathog. 2014. PMID: 25033463 Free PMC article.
-
ORF45-Mediated Prolonged c-Fos Accumulation Accelerates Viral Transcription during the Late Stage of Lytic Replication of Kaposi's Sarcoma-Associated Herpesvirus.J Virol. 2015 Jul;89(13):6895-906. doi: 10.1128/JVI.00274-15. Epub 2015 Apr 22. J Virol. 2015. PMID: 25903346 Free PMC article.
-
The open chromatin landscape of Kaposi's sarcoma-associated herpesvirus.J Virol. 2013 Nov;87(21):11831-42. doi: 10.1128/JVI.01685-13. Epub 2013 Aug 28. J Virol. 2013. PMID: 23986576 Free PMC article.
-
Rapid deamination of cyclobutane pyrimidine dimer photoproducts at TCG sites in a translationally and rotationally positioned nucleosome in vivo.J Biol Chem. 2015 Oct 30;290(44):26597-609. doi: 10.1074/jbc.M115.673301. Epub 2015 Sep 9. J Biol Chem. 2015. PMID: 26354431 Free PMC article.
-
Kaposi's Sarcoma-Associated Herpesvirus Hijacks RNA Polymerase II To Create a Viral Transcriptional Factory.J Virol. 2017 May 12;91(11):e02491-16. doi: 10.1128/JVI.02491-16. Print 2017 Jun 1. J Virol. 2017. PMID: 28331082 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
