

Viral quasispecies evolution
- PMID: 22688811
- PMCID: PMC3372249
- DOI: 10.1128/MMBR.05023-11
Viral quasispecies evolution
Abstract
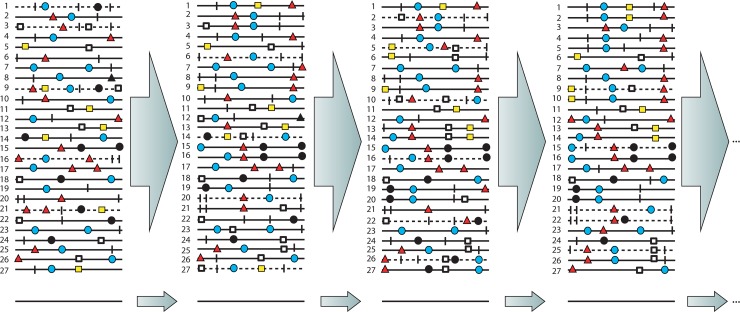
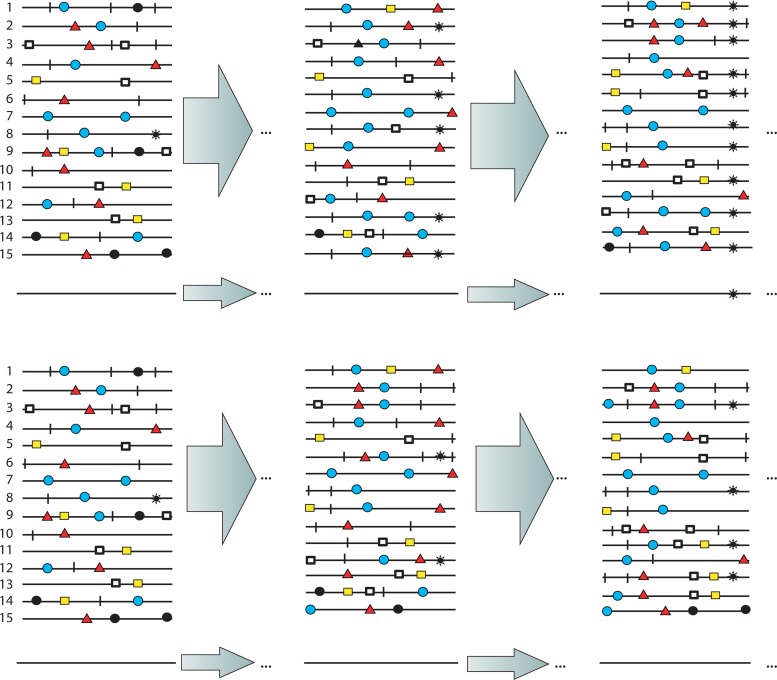
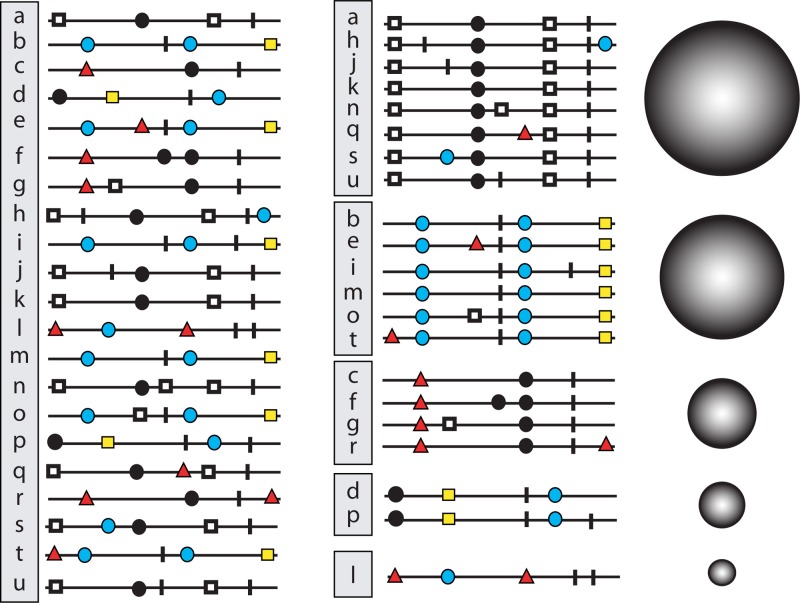
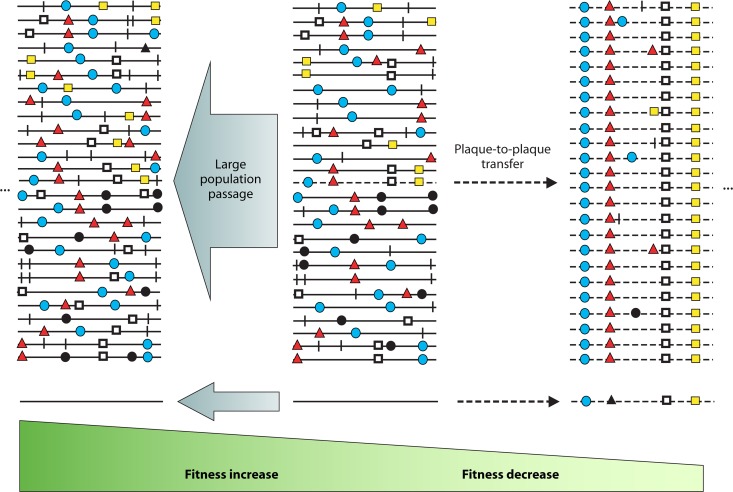
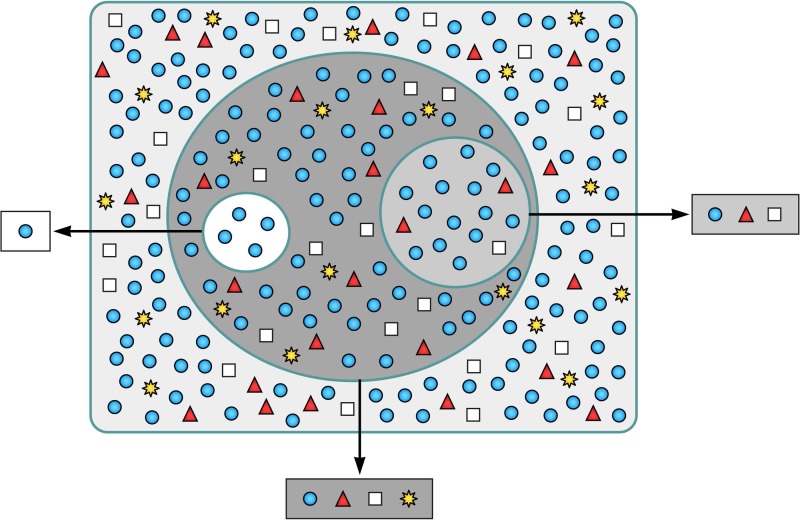
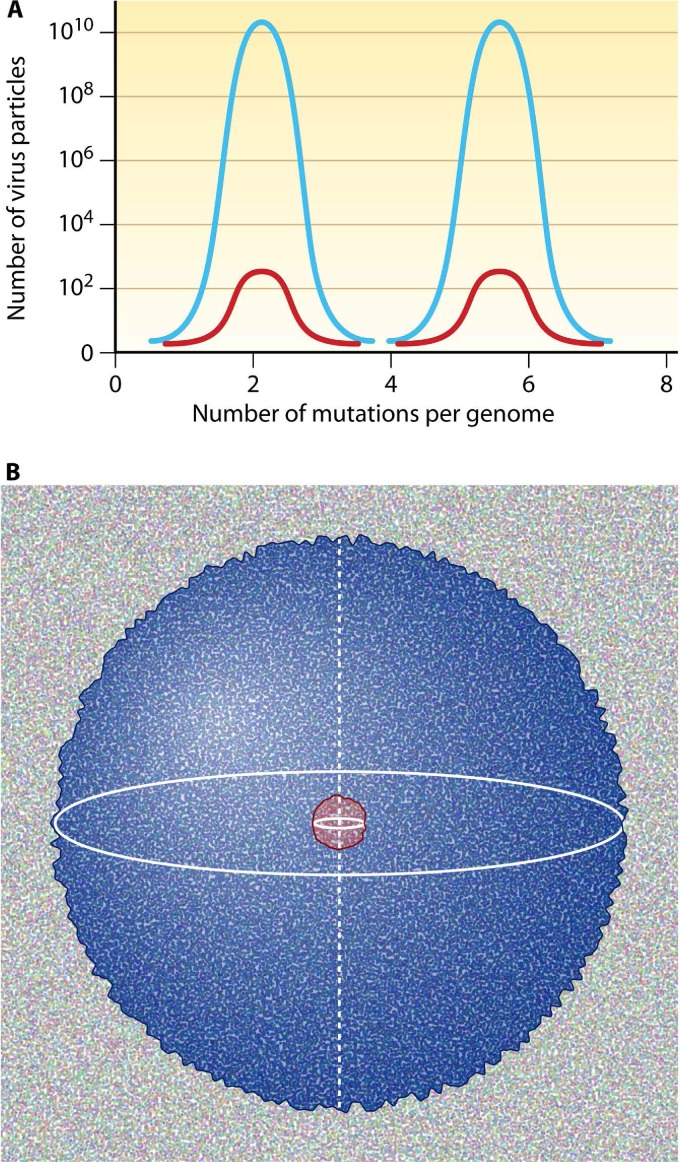
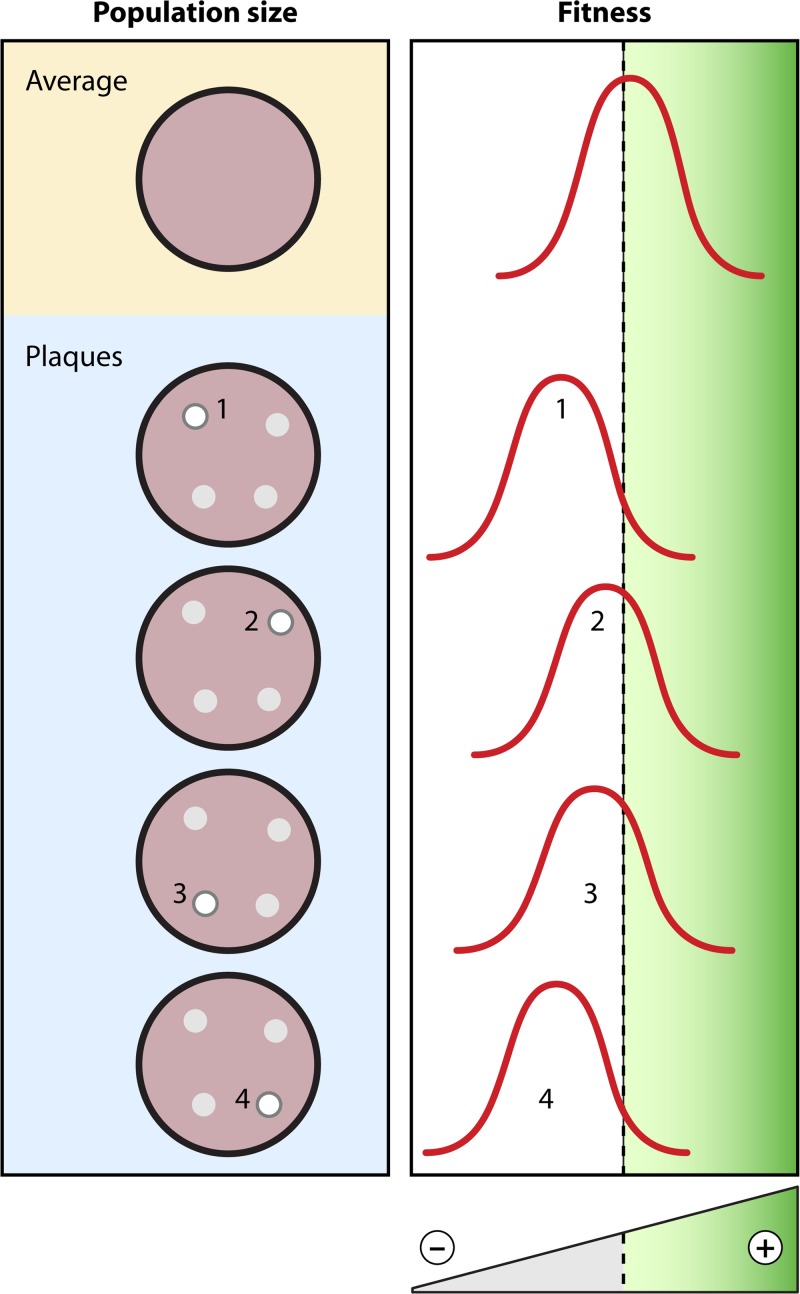
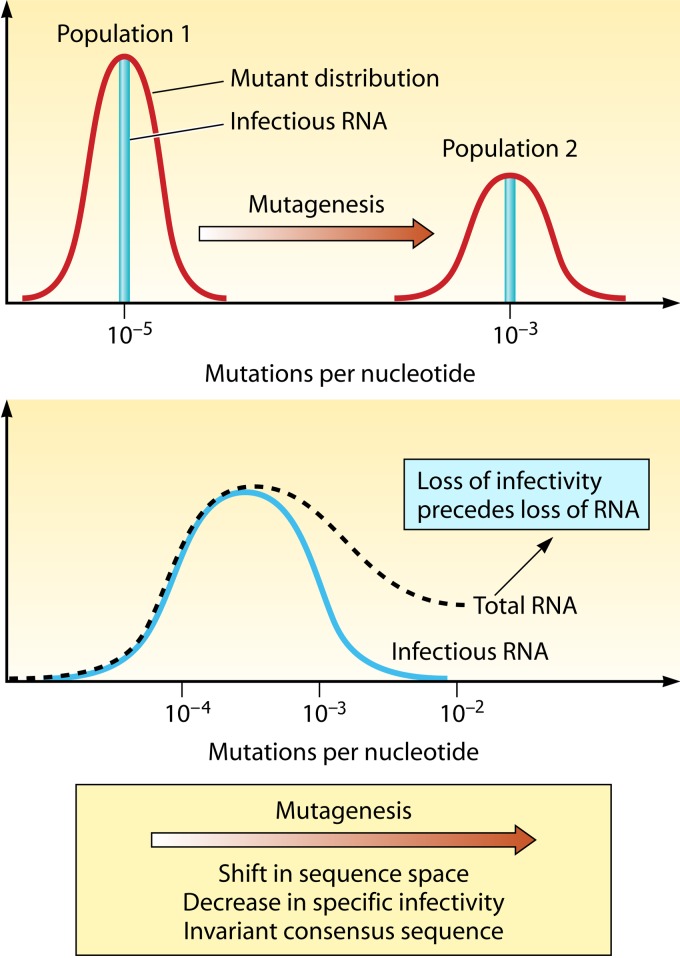
Evolution of RNA viruses occurs through disequilibria of collections of closely related mutant spectra or mutant clouds termed viral quasispecies. Here we review the origin of the quasispecies concept and some biological implications of quasispecies dynamics. Two main aspects are addressed: (i) mutant clouds as reservoirs of phenotypic variants for virus adaptability and (ii) the internal interactions that are established within mutant spectra that render a virus ensemble the unit of selection. The understanding of viruses as quasispecies has led to new antiviral designs, such as lethal mutagenesis, whose aim is to drive viruses toward low fitness values with limited chances of fitness recovery. The impact of quasispecies for three salient human pathogens, human immunodeficiency virus and the hepatitis B and C viruses, is reviewed, with emphasis on antiviral treatment strategies. Finally, extensions of quasispecies to nonviral systems are briefly mentioned to emphasize the broad applicability of quasispecies theory.
Figures
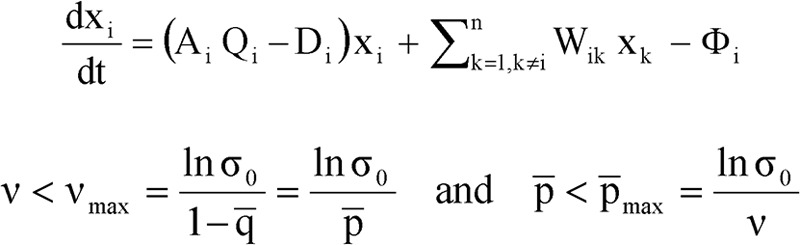




Similar articles
-
Unfinished stories on viral quasispecies and Darwinian views of evolution.J Mol Biol. 2010 Apr 9;397(4):865-77. doi: 10.1016/j.jmb.2010.02.005. Epub 2010 Feb 10. J Mol Biol. 2010. PMID: 20152841 Review.
-
Quasispecies and the development of new antiviral strategies.Prog Drug Res. 2003;60:133-58. doi: 10.1007/978-3-0348-8012-1_4. Prog Drug Res. 2003. PMID: 12790341 Review.
-
Viral quasispecies.PLoS Genet. 2019 Oct 17;15(10):e1008271. doi: 10.1371/journal.pgen.1008271. eCollection 2019 Oct. PLoS Genet. 2019. PMID: 31622336 Free PMC article.
-
RNA virus quasispecies: significance for viral disease and epidemiology.Infect Agents Dis. 1994 Aug;3(4):201-14. Infect Agents Dis. 1994. PMID: 7827789 Review.
-
Historical Perspective on the Discovery of the Quasispecies Concept.Annu Rev Virol. 2021 Sep 29;8(1):51-72. doi: 10.1146/annurev-virology-091919-105900. Annu Rev Virol. 2021. PMID: 34586874 Review.
Cited by
-
Role of cross-reactivity in cellular immune targeting of influenza A M158-66 variant peptide epitopes.Front Immunol. 2022 Sep 23;13:956103. doi: 10.3389/fimmu.2022.956103. eCollection 2022. Front Immunol. 2022. PMID: 36211433 Free PMC article.
-
Phylogenetic analysis of the neuraminidase segment gene of Influenza A/H1N1 strains isolated from Monastir Region (Tunisia) during the 2017-2018 outbreak.Biologia (Bratisl). 2021;76(6):1797-1806. doi: 10.1007/s11756-021-00723-y. Epub 2021 Mar 12. Biologia (Bratisl). 2021. PMID: 33727729 Free PMC article.
-
Response of hepatitis C virus to long-term passage in the presence of alpha interferon: multiple mutations and a common phenotype.J Virol. 2013 Jul;87(13):7593-607. doi: 10.1128/JVI.02824-12. Epub 2013 May 1. J Virol. 2013. PMID: 23637397 Free PMC article.
-
Crucial mutation in the exoribonuclease domain of nsp14 of PEDV leads to high genetic instability during viral replication.Cell Biosci. 2021 Jun 7;11(1):106. doi: 10.1186/s13578-021-00598-1. Cell Biosci. 2021. PMID: 34099051 Free PMC article.
-
The Role of Receptor Tyrosine Kinases in Lassa Virus Cell Entry.Viruses. 2020 Aug 6;12(8):857. doi: 10.3390/v12080857. Viruses. 2020. PMID: 32781509 Free PMC article.
References
-
- Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. 2006. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 311:236–238 - PubMed
-
- Acharya R, et al. 1989. The three-dimensional structure of foot-and-mouth disease virus at 2.9 Å resolution. Nature 337:709–716 - PubMed
-
- Agol VI. 2006. Molecular mechanisms of poliovirus variation and evolution. Curr. Top. Microbiol. Immunol. 299:211–259 - PubMed
-
- Agol VI. 2010. Picornaviruses as a model for studying the nature of RNA recombination, p 239–252 In Ehrenfeld E, Domingo E, Roos RP. (ed), The Picornaviruses. ASM Press, Washington, DC
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
