

Discovery of small molecule cancer drugs: successes, challenges and opportunities
- PMID: 22440008
- PMCID: PMC3476506
- DOI: 10.1016/j.molonc.2012.02.004
Discovery of small molecule cancer drugs: successes, challenges and opportunities
Abstract
The discovery and development of small molecule cancer drugs has been revolutionised over the last decade. Most notably, we have moved from a one-size-fits-all approach that emphasized cytotoxic chemotherapy to a personalised medicine strategy that focuses on the discovery and development of molecularly targeted drugs that exploit the particular genetic addictions, dependencies and vulnerabilities of cancer cells. These exploitable characteristics are increasingly being revealed by our expanding understanding of the abnormal biology and genetics of cancer cells, accelerated by cancer genome sequencing and other high-throughput genome-wide campaigns, including functional screens using RNA interference. In this review we provide an overview of contemporary approaches to the discovery of small molecule cancer drugs, highlighting successes, current challenges and future opportunities. We focus in particular on four key steps: Target validation and selection; chemical hit and lead generation; lead optimization to identify a clinical drug candidate; and finally hypothesis-driven, biomarker-led clinical trials. Although all of these steps are critical, we view target validation and selection and the conduct of biology-directed clinical trials as especially important areas upon which to focus to speed progress from gene to drug and to reduce the unacceptably high attrition rate during clinical development. Other challenges include expanding the envelope of druggability for less tractable targets, understanding and overcoming drug resistance, and designing intelligent and effective drug combinations. We discuss not only scientific and technical challenges, but also the assessment and mitigation of risks as well as organizational, cultural and funding problems for cancer drug discovery and development, together with solutions to overcome the 'Valley of Death' between basic research and approved medicines. We envisage a future in which addressing these challenges will enhance our rapid progress towards truly personalised medicine for cancer patients.
Copyright © 2012 Federation of European Biochemical Societies. Published by Elsevier B.V. All rights reserved.
Figures
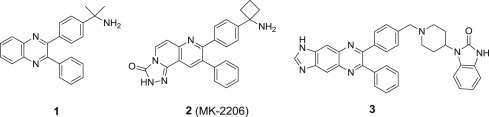
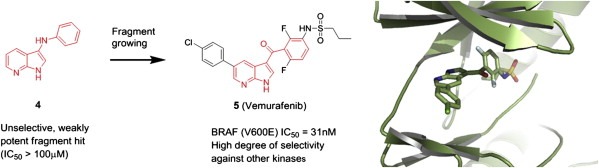
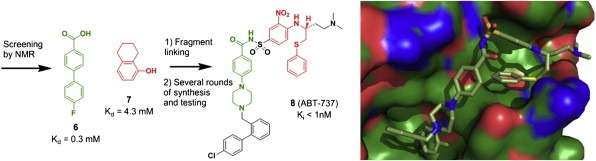
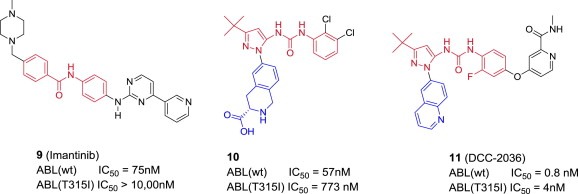
Similar articles
-
Exploiting the cancer genome: strategies for the discovery and clinical development of targeted molecular therapeutics.Annu Rev Pharmacol Toxicol. 2012;52:549-73. doi: 10.1146/annurev-pharmtox-010611-134532. Annu Rev Pharmacol Toxicol. 2012. PMID: 22235862 Review.
-
The opportunities and challenges of personalized genome-based molecular therapies for cancer: targets, technologies, and molecular chaperones.Cancer Chemother Pharmacol. 2003 Jul;52 Suppl 1:S45-56. doi: 10.1007/s00280-003-0593-0. Epub 2003 Jun 18. Cancer Chemother Pharmacol. 2003. PMID: 12819933 Review.
-
Overview on the current status of virtual high-throughput screening and combinatorial chemistry approaches in multi-target anticancer drug discovery; Part I.J BUON. 2016 Jul-Aug;21(4):764-779. J BUON. 2016. PMID: 27685895 Review.
-
New tools for old drugs: Functional genetic screens to optimize current chemotherapy.Drug Resist Updat. 2018 Jan;36:30-46. doi: 10.1016/j.drup.2018.01.001. Epub 2018 Jan 12. Drug Resist Updat. 2018. PMID: 29499836 Free PMC article. Review.
-
[Development of antituberculous drugs: current status and future prospects].Kekkaku. 2006 Dec;81(12):753-74. Kekkaku. 2006. PMID: 17240921 Review. Japanese.
Cited by
-
iGMDR: Integrated Pharmacogenetic Resource Guide to Cancer Therapy and Research.Genomics Proteomics Bioinformatics. 2020 Apr;18(2):150-160. doi: 10.1016/j.gpb.2019.11.011. Epub 2020 Sep 8. Genomics Proteomics Bioinformatics. 2020. PMID: 32916316 Free PMC article.
-
Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways.Mol Cancer. 2022 Oct 12;21(1):196. doi: 10.1186/s12943-022-01664-z. Mol Cancer. 2022. PMID: 36221123 Free PMC article. Review.
-
Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery.Int J Mol Sci. 2020 Jul 24;21(15):5262. doi: 10.3390/ijms21155262. Int J Mol Sci. 2020. PMID: 32722222 Free PMC article. Review.
-
The SHREAD gene therapy platform for paracrine delivery improves tumor localization and intratumoral effects of a clinical antibody.Proc Natl Acad Sci U S A. 2021 May 25;118(21):e2017925118. doi: 10.1073/pnas.2017925118. Proc Natl Acad Sci U S A. 2021. PMID: 34001602 Free PMC article.
-
MicroRNA-146a limits tumorigenic inflammation in colorectal cancer.Nat Commun. 2021 Apr 23;12(1):2419. doi: 10.1038/s41467-021-22641-y. Nat Commun. 2021. PMID: 33893298 Free PMC article.
References
-
- Abad-Zapatero, C. , Metz, J.T. , 2005. Ligand efficiency indices as guideposts for drug discovery. Drug Discov. Today. 10, 464–469. - PubMed
-
- Akella, L.B. , DeCaprio, D. , 2010. Cheminformatics approaches to analyze diversity in compound screening libraries. Curr. Opin. Chem. Biol.. 14, 325–330. - PubMed
-
- Allegra, C.J. , Jessup, J.M. , Somerfield, M.R. , Hamilton, S.R. , Hammond, E.H. , Hayes, D.F. , McAllister, P.K. , Morton, R.F. , Schilsky, R.L. , 2009. American society of clinical oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol.. 27, 2091–2096. - PubMed
-
- Ashworth, A. , Lord, Christopher J. , Reis-Filho, Jorge S. , 2011. Genetic interactions in cancer progression and treatment. Cell. 145, 30–38. - PubMed
-
- Baell, J.B. , Holloway, G.E. , 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem.. 53, 2719–2740. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
