

Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses
- PMID: 22067224
- PMCID: PMC3228705
- DOI: 10.1186/1742-4690-8-90
Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses
Abstract
Background: Integration of retroviral DNA into a germ cell may lead to a provirus that is transmitted vertically to that host's offspring as an endogenous retrovirus (ERV). In humans, ERVs (HERVs) comprise about 8% of the genome, the vast majority of which are truncated and/or highly mutated and no longer encode functional genes. The most recently active retroviruses that integrated into the human germ line are members of the Betaretrovirus-like HERV-K (HML-2) group, many of which contain intact open reading frames (ORFs) in some or all genes, sometimes encoding functional proteins that are expressed in various tissues. Interestingly, this expression is upregulated in many tumors ranging from breast and ovarian tissues to lymphomas and melanomas, as well as schizophrenia, rheumatoid arthritis, and other disorders.
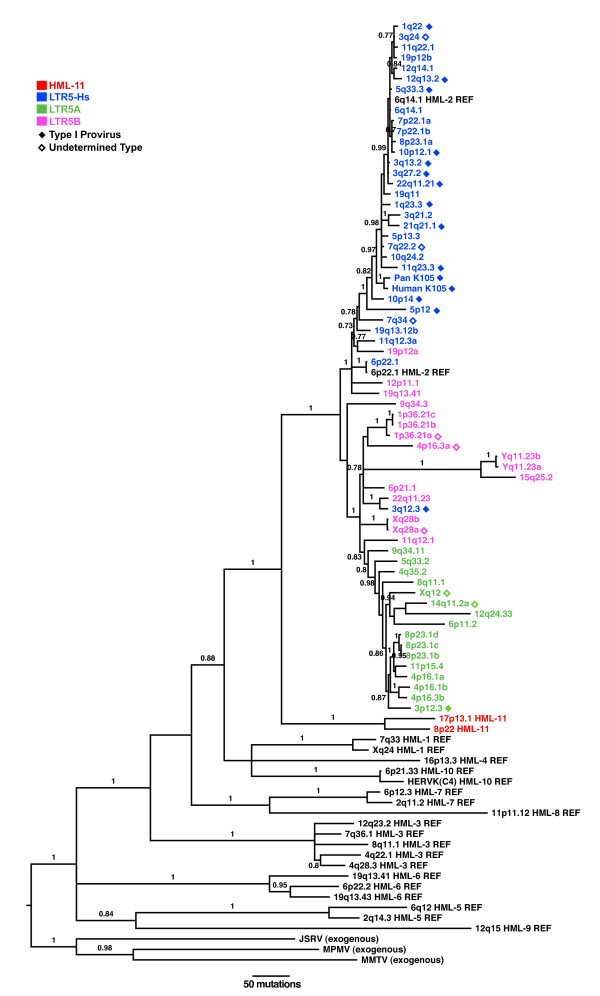
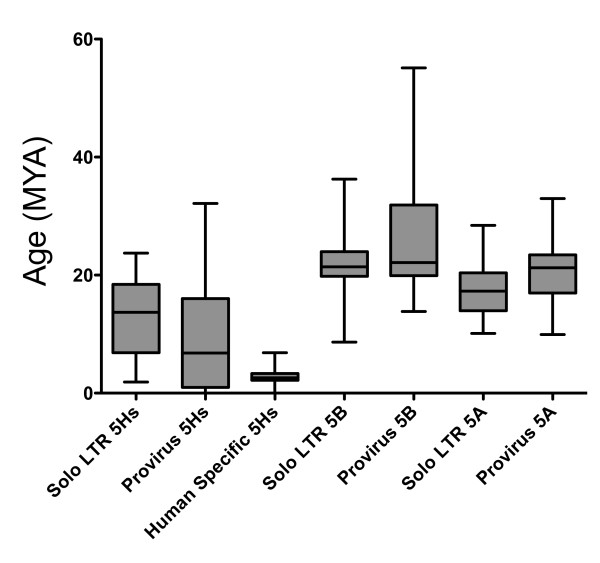
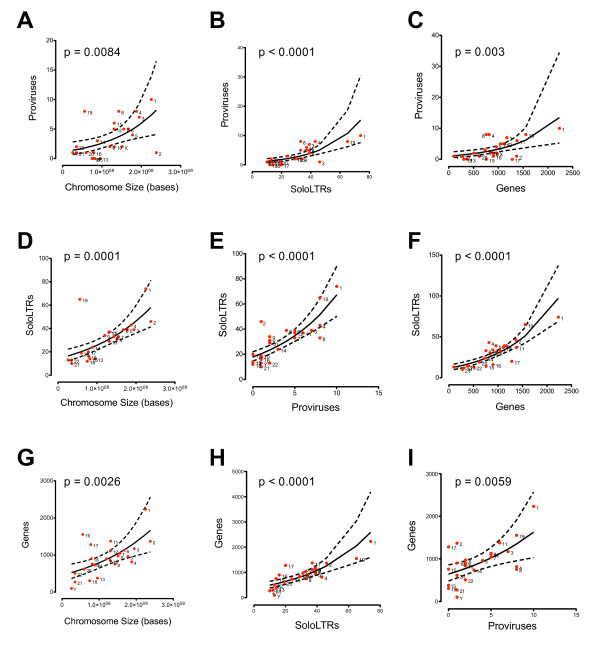
Results: No study to date has characterized all HML-2 elements in the genome, an essential step towards determining a possible functional role of HML-2 expression in disease. We present here the most comprehensive and accurate catalog of all full-length and partial HML-2 proviruses, as well as solo LTR elements, within the published human genome to date. Furthermore, we provide evidence for preferential maintenance of proviruses and solo LTR elements on gene-rich chromosomes of the human genome and in proximity to gene regions.
Conclusions: Our analysis has found and corrected several errors in the annotation of HML-2 elements in the human genome, including mislabeling of a newly identified group called HML-11. HML-elements have been implicated in a wide array of diseases, and characterization of these elements will play a fundamental role to understand the relationship between endogenous retrovirus expression and disease.
Figures



Similar articles
-
Comprehensive Characterization of the Human Endogenous Retrovirus HERV-K(HML-6) Group: Overview of Structure, Phylogeny, and Contribution to the Human Genome.J Virol. 2019 Jul 30;93(16):e00110-19. doi: 10.1128/JVI.00110-19. Print 2019 Aug 15. J Virol. 2019. PMID: 31167914 Free PMC article.
-
Genome-wide amplification of proviral sequences reveals new polymorphic HERV-K(HML-2) proviruses in humans and chimpanzees that are absent from genome assemblies.Retrovirology. 2015 Apr 28;12:35. doi: 10.1186/s12977-015-0162-8. Retrovirology. 2015. PMID: 25927962 Free PMC article.
-
Human endogenous retrovirus family HERV-K(HML-5): status, evolution, and reconstruction of an ancient betaretrovirus in the human genome.J Virol. 2004 Aug;78(16):8788-98. doi: 10.1128/JVI.78.16.8788-8798.2004. J Virol. 2004. PMID: 15280487 Free PMC article.
-
Human endogenous retrovirus-K (HML-2): a comprehensive review.Crit Rev Microbiol. 2018 Nov;44(6):715-738. doi: 10.1080/1040841X.2018.1501345. Epub 2018 Oct 14. Crit Rev Microbiol. 2018. PMID: 30318978 Free PMC article. Review.
-
Human Endogenous Retrovirus-K (HML-2)-Related Genetic Variation: Human Genome Diversity and Disease.Genes (Basel). 2023 Nov 28;14(12):2150. doi: 10.3390/genes14122150. Genes (Basel). 2023. PMID: 38136972 Free PMC article. Review.
Cited by
-
Human Endogenous Retrovirus K in Cancer: A Potential Biomarker and Immunotherapeutic Target.Viruses. 2020 Jul 6;12(7):726. doi: 10.3390/v12070726. Viruses. 2020. PMID: 32640516 Free PMC article. Review.
-
HERV-K-specific T cells eliminate diverse HIV-1/2 and SIV primary isolates.J Clin Invest. 2012 Dec;122(12):4473-89. doi: 10.1172/JCI64560. Epub 2012 Nov 12. J Clin Invest. 2012. PMID: 23143309 Free PMC article.
-
Integration and Fixation Preferences of Human and Mouse Endogenous Retroviruses Uncovered with Functional Data Analysis.PLoS Comput Biol. 2016 Jun 16;12(6):e1004956. doi: 10.1371/journal.pcbi.1004956. eCollection 2016 Jun. PLoS Comput Biol. 2016. PMID: 27309962 Free PMC article.
-
Structural basis for Fullerene geometry in a human endogenous retrovirus capsid.Nat Commun. 2019 Dec 20;10(1):5822. doi: 10.1038/s41467-019-13786-y. Nat Commun. 2019. PMID: 31862888 Free PMC article.
-
Human endogenous retrovirus K in the respiratory tract is associated with COVID-19 physiopathology.Microbiome. 2022 Apr 22;10(1):65. doi: 10.1186/s40168-022-01260-9. Microbiome. 2022. PMID: 35459226 Free PMC article.
References
-
- Boeke JD, Stoye JP. In: Retroviruses. Coffin JM, Hughes SH, Varmus HE, editor. Cold Spring Harbor Laboratory Press; 1997. Retrotransposons, Endogenous Retroviruses, and the Evolution of Retroelements; pp. 343–436. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
