

Comparative analysis of proteome and transcriptome variation in mouse
- PMID: 21695224
- PMCID: PMC3111477
- DOI: 10.1371/journal.pgen.1001393
Comparative analysis of proteome and transcriptome variation in mouse
Abstract
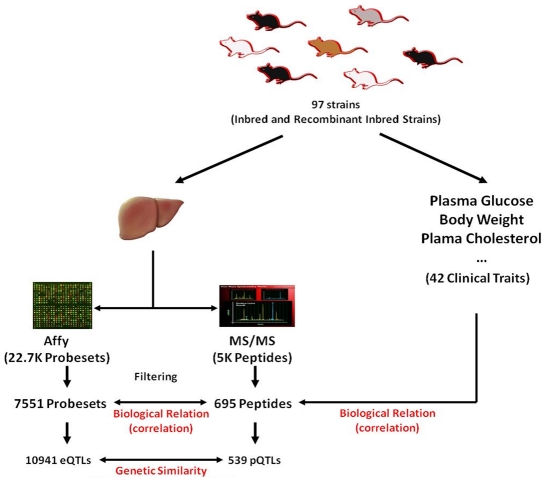
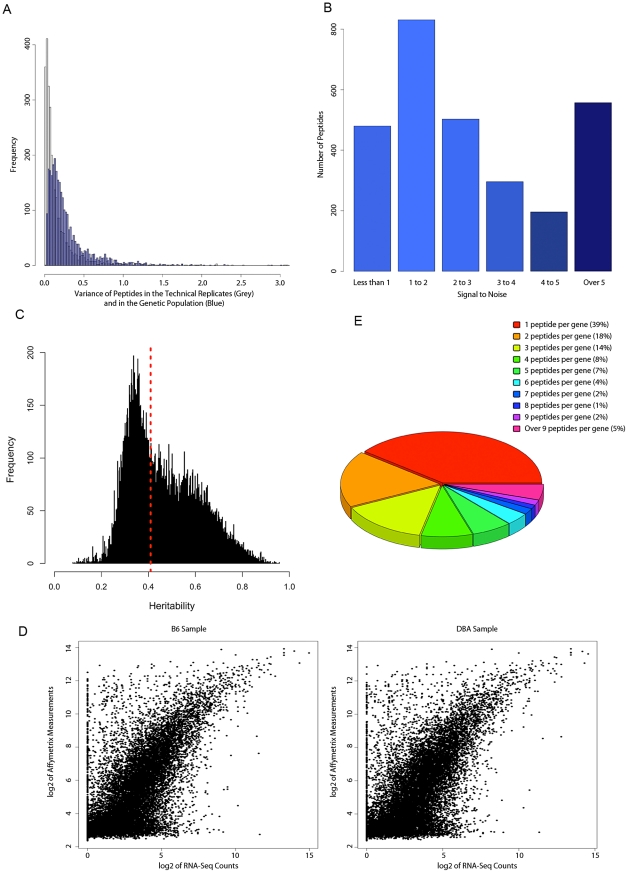
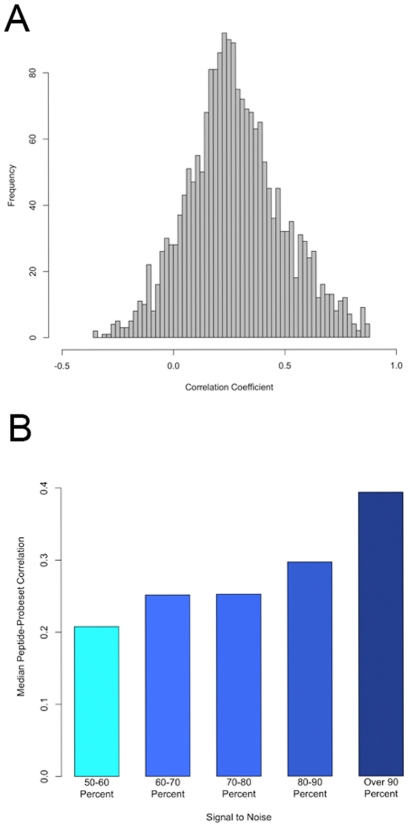
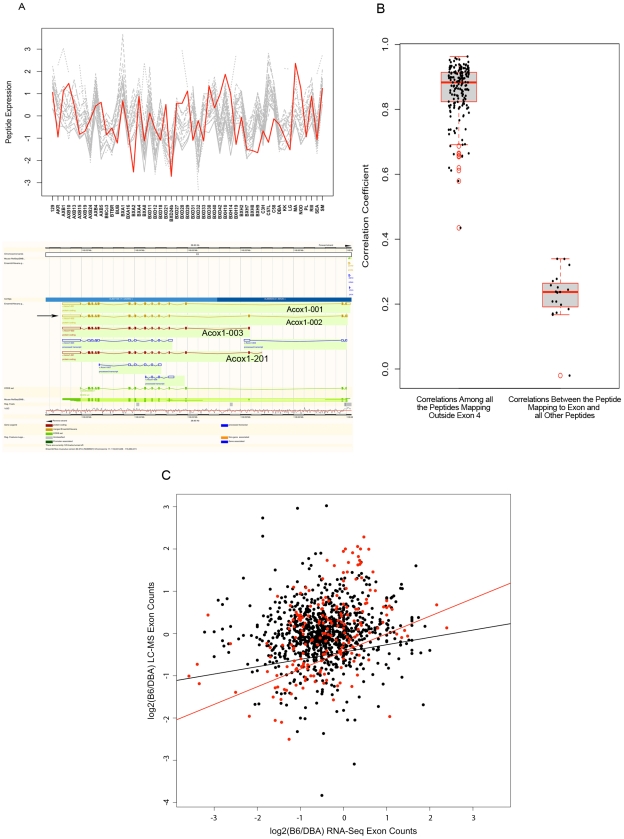
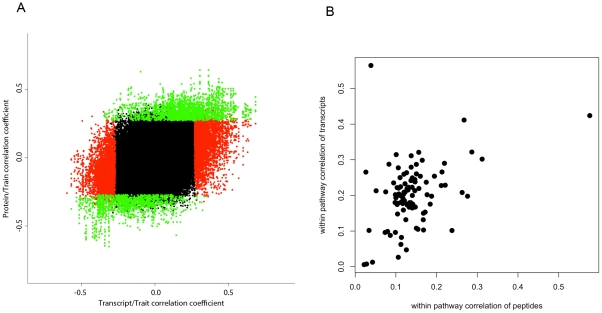
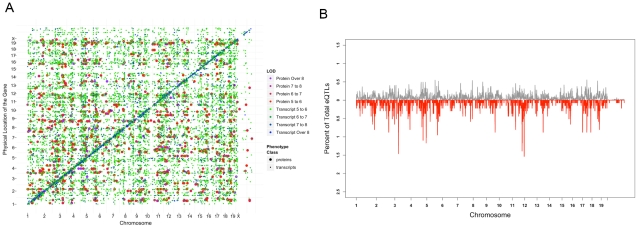
The relationships between the levels of transcripts and the levels of the proteins they encode have not been examined comprehensively in mammals, although previous work in plants and yeast suggest a surprisingly modest correlation. We have examined this issue using a genetic approach in which natural variations were used to perturb both transcript levels and protein levels among inbred strains of mice. We quantified over 5,000 peptides and over 22,000 transcripts in livers of 97 inbred and recombinant inbred strains and focused on the 7,185 most heritable transcripts and 486 most reliable proteins. The transcript levels were quantified by microarray analysis in three replicates and the proteins were quantified by Liquid Chromatography-Mass Spectrometry using O(18)-reference-based isotope labeling approach. We show that the levels of transcripts and proteins correlate significantly for only about half of the genes tested, with an average correlation of 0.27, and the correlations of transcripts and proteins varied depending on the cellular location and biological function of the gene. We examined technical and biological factors that could contribute to the modest correlation. For example, differential splicing clearly affects the analyses for certain genes; but, based on deep sequencing, this does not substantially contribute to the overall estimate of the correlation. We also employed genome-wide association analyses to map loci controlling both transcript and protein levels. Surprisingly, little overlap was observed between the protein- and transcript-mapped loci. We have typed numerous clinically relevant traits among the strains, including adiposity, lipoprotein levels, and tissue parameters. Using correlation analysis, we found that a low number of clinical trait relationships are preserved between the protein and mRNA gene products and that the majority of such relationships are specific to either the protein levels or transcript levels. Surprisingly, transcript levels were more strongly correlated with clinical traits than protein levels. In light of the widespread use of high-throughput technologies in both clinical and basic research, the results presented have practical as well as basic implications.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
Gene expression: Transcriptome to proteome and back to genome.Nat Rev Genet. 2011 Jun 28;12(8):518. doi: 10.1038/nrg3037. Nat Rev Genet. 2011. PMID: 21709688 No abstract available.
Similar articles
-
Transcriptomic and proteomic profiling of two porcine tissues using high-throughput technologies.BMC Genomics. 2009 Jan 19;10:30. doi: 10.1186/1471-2164-10-30. BMC Genomics. 2009. PMID: 19152685 Free PMC article.
-
Defining the consequences of genetic variation on a proteome-wide scale.Nature. 2016 Jun 23;534(7608):500-5. doi: 10.1038/nature18270. Epub 2016 Jun 15. Nature. 2016. PMID: 27309819 Free PMC article.
-
Variation and genetic control of protein abundance in humans.Nature. 2013 Jul 4;499(7456):79-82. doi: 10.1038/nature12223. Epub 2013 May 15. Nature. 2013. PMID: 23676674 Free PMC article.
-
Workability of mRNA Sequencing for Predicting Protein Abundance.Genes (Basel). 2023 Nov 11;14(11):2065. doi: 10.3390/genes14112065. Genes (Basel). 2023. PMID: 38003008 Free PMC article. Review.
-
Genome stability versus transcript diversity.DNA Repair (Amst). 2016 Aug;44:81-86. doi: 10.1016/j.dnarep.2016.05.010. Epub 2016 May 16. DNA Repair (Amst). 2016. PMID: 27246512 Free PMC article. Review.
Cited by
-
Nicotinic Receptor Subunit Distribution in Auditory Cortex: Impact of Aging on Receptor Number and Function.J Neurosci. 2020 Jul 22;40(30):5724-5739. doi: 10.1523/JNEUROSCI.0093-20.2020. Epub 2020 Jun 15. J Neurosci. 2020. PMID: 32541068 Free PMC article.
-
Integration of omic networks in a developmental atlas of maize.Science. 2016 Aug 19;353(6301):814-8. doi: 10.1126/science.aag1125. Science. 2016. PMID: 27540173 Free PMC article.
-
Bioinformatics and HIV latency.Curr HIV/AIDS Rep. 2015 Mar;12(1):97-106. doi: 10.1007/s11904-014-0240-x. Curr HIV/AIDS Rep. 2015. PMID: 25586146 Free PMC article. Review.
-
Pressure-overload hypertrophy of the developing heart reveals activation of divergent gene and protein pathways in the left and right ventricular myocardium.Am J Physiol Heart Circ Physiol. 2013 Mar 1;304(5):H697-708. doi: 10.1152/ajpheart.00802.2012. Epub 2012 Dec 21. Am J Physiol Heart Circ Physiol. 2013. PMID: 23262132 Free PMC article.
-
TRIM28 Is a Novel Regulator of CD133 Expression Associated with Cancer Stem Cell Phenotype.Int J Mol Sci. 2022 Aug 30;23(17):9874. doi: 10.3390/ijms23179874. Int J Mol Sci. 2022. PMID: 36077272 Free PMC article.
References
-
- Brem RB, Yvert G, Clinton R, Kruglyak L. Genetic dissection of transcriptional regulation in budding yeast. Science. 2002;296:752–755. - PubMed
-
- Bystrykh L, Weersing E, Dontje B, Sutton S, Pletcher MT, et al. Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat Genet. 2005;37:225–232. - PubMed
-
- Ghazalpour A, Doss S, Kang H, Farber C, Wen PZ, et al. High-resolution mapping of gene expression using association in an outbred mouse stock. PLoS Genet. 2008;4:e1000149. doi: 10.1371/journal.pgen.1000149. - DOI - PMC - PubMed
-
- Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2:e130. doi: 10.1371/journal.pgen.0020130. - DOI - PMC - PubMed
-
- Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, et al. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet. 2005;37:243–253. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL094322/HL/NHLBI NIH HHS/United States
- T32-HG002536/HG/NHGRI NIH HHS/United States
- R00 HL094709/HL/NHLBI NIH HHS/United States
- GM07104/GM/NIGMS NIH HHS/United States
- HL28481/HL/NHLBI NIH HHS/United States
- T32 HG002536/HG/NHGRI NIH HHS/United States
- K25 HL080079/HL/NHLBI NIH HHS/United States
- 5F32DK074317/DK/NIDDK NIH HHS/United States
- T32 GM007104/GM/NIGMS NIH HHS/United States
- HL30568/HL/NHLBI NIH HHS/United States
- F32 DK074317/DK/NIDDK NIH HHS/United States
- AHA0825204F/PHS HHS/United States
- K99 HL102223/HL/NHLBI NIH HHS/United States
- RR18552/RR/NCRR NIH HHS/United States
- R01 HL094322/HL/NHLBI NIH HHS/United States
- R01 NS050148/NS/NINDS NIH HHS/United States
- P01 HL028481/HL/NHLBI NIH HHS/United States
- K99 HL102223-01A1/HL/NHLBI NIH HHS/United States
- P01 HL030568/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
