

The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones
- PMID: 21228324
- PMCID: PMC3062313
- DOI: 10.1182/blood-2010-10-312926
The host genomic environment of the provirus determines the abundance of HTLV-1-infected T-cell clones
Abstract
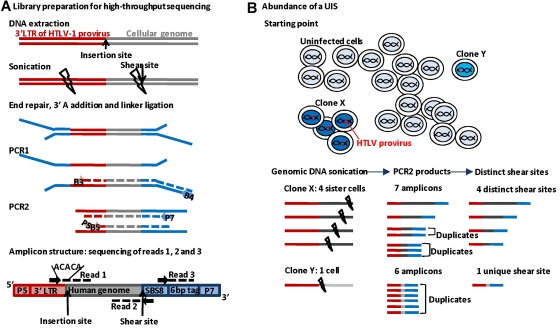
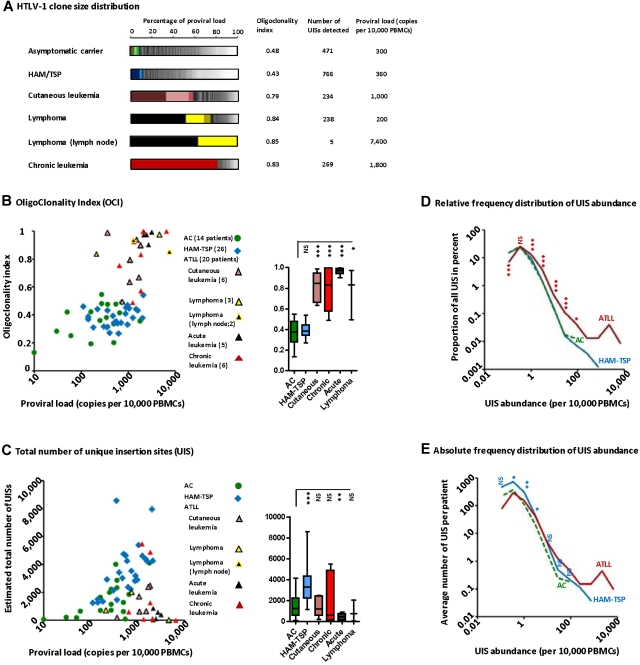
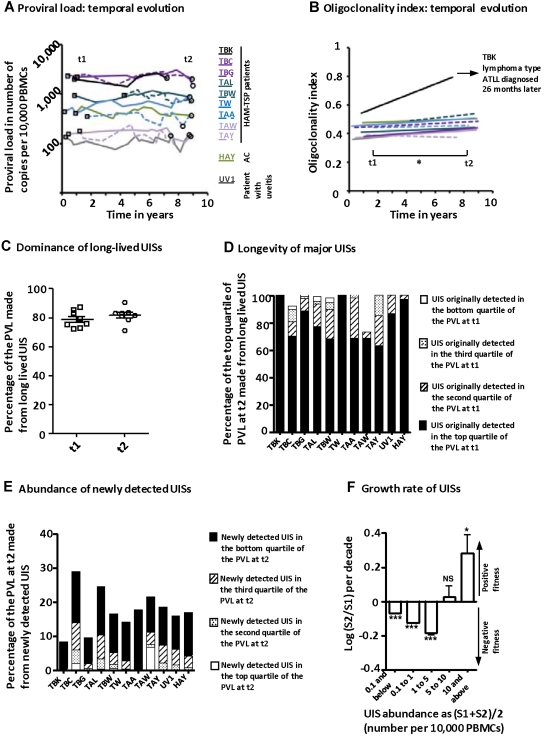
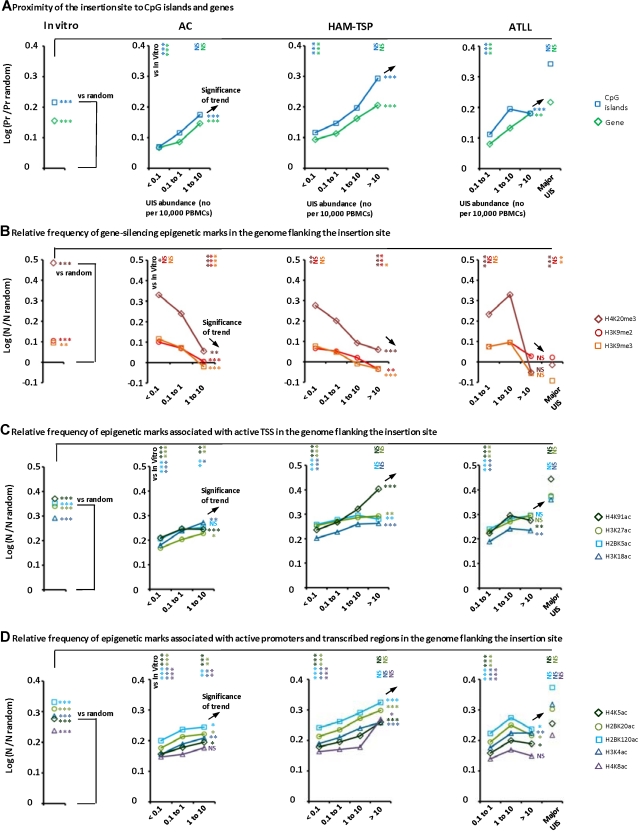
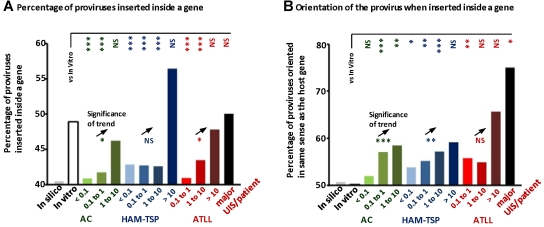
Human T-lymphotropic virus type 1 (HTLV-1) persists by driving clonal proliferation of infected T lymphocytes. A high proviral load predisposes to HTLV-1-associated diseases. Yet the reasons for the variation within and between persons in the abundance of HTLV-1-infected clones remain unknown. We devised a high-throughput protocol to map the genomic location and quantify the abundance of > 91,000 unique insertion sites of the provirus from 61 HTLV-1(+) persons and > 2100 sites from in vitro infection. We show that a typical HTLV-1-infected host carries between 500 and 5000 unique insertion sites. We demonstrate that negative selection dominates during chronic infection, favoring establishment of proviruses integrated in transcriptionally silenced DNA: this selection is significantly stronger in asymptomatic carriers. We define a parameter, the oligoclonality index, to quantify clonality. The high proviral load characteristic of HTLV-1-associated inflammatory disease results from a larger number of unique insertion sites than in asymptomatic carriers and not, as previously thought, from a difference in clonality. The abundance of established HTLV-1 clones is determined by genomic features of the host DNA flanking the provirus. HTLV-1 clonal expansion in vivo is favored by orientation of the provirus in the same sense as the nearest host gene.
Figures
Similar articles
-
The role of HTLV-1 clonality, proviral structure, and genomic integration site in adult T-cell leukemia/lymphoma.Blood. 2014 Jun 19;123(25):3925-31. doi: 10.1182/blood-2014-02-553602. Epub 2014 Apr 15. Blood. 2014. PMID: 24735963 Free PMC article.
-
The retrovirus HTLV-1 inserts an ectopic CTCF-binding site into the human genome.Proc Natl Acad Sci U S A. 2016 Mar 15;113(11):3054-9. doi: 10.1073/pnas.1423199113. Epub 2016 Feb 29. Proc Natl Acad Sci U S A. 2016. PMID: 26929370 Free PMC article.
-
Strongyloidiasis and infective dermatitis alter human T lymphotropic virus-1 clonality in vivo.PLoS Pathog. 2013;9(4):e1003263. doi: 10.1371/journal.ppat.1003263. Epub 2013 Apr 4. PLoS Pathog. 2013. PMID: 23592987 Free PMC article.
-
HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection.Semin Cancer Biol. 2014 Jun;26:89-98. doi: 10.1016/j.semcancer.2013.11.003. Epub 2013 Dec 6. Semin Cancer Biol. 2014. PMID: 24316494 Free PMC article. Review.
-
Transcriptional and Epigenetic Regulatory Mechanisms Affecting HTLV-1 Provirus.Viruses. 2016 Jun 16;8(6):171. doi: 10.3390/v8060171. Viruses. 2016. PMID: 27322309 Free PMC article. Review.
Cited by
-
HTLV-1-infected T cells contain a single integrated provirus in natural infection.Blood. 2012 Oct 25;120(17):3488-90. doi: 10.1182/blood-2012-07-445593. Epub 2012 Sep 6. Blood. 2012. PMID: 22955925 Free PMC article.
-
Tuning Rex rules HTLV-1 pathogenesis.Front Immunol. 2022 Sep 16;13:959962. doi: 10.3389/fimmu.2022.959962. eCollection 2022. Front Immunol. 2022. PMID: 36189216 Free PMC article. Review.
-
Retrieval of vector integration sites from cell-free DNA.Nat Med. 2021 Aug;27(8):1458-1470. doi: 10.1038/s41591-021-01389-4. Epub 2021 Jun 17. Nat Med. 2021. PMID: 34140705
-
Bioinformatic clonality analysis of next-generation sequencing-derived viral vector integration sites.Hum Gene Ther Methods. 2012 Apr;23(2):111-8. doi: 10.1089/hgtb.2011.219. Epub 2012 May 4. Hum Gene Ther Methods. 2012. PMID: 22559057 Free PMC article.
-
The forces driving clonal expansion of the HIV-1 latent reservoir.Virol J. 2020 Jan 7;17(1):4. doi: 10.1186/s12985-019-1276-8. Virol J. 2020. PMID: 31910871 Free PMC article. Review.
References
-
- Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene. 2005;24(39):6058–6068. - PubMed
-
- Kubota R, Fujiyoshi T, Izumo S, et al. Fluctuation of HTLV-I proviral DNA in peripheral blood mononuclear cells of HTLV-I-associated myelopathy. J Neuroimmunol. 1993;42(2):147–154. - PubMed
-
- Nagai M, Usuku K, Matsumoto W, et al. Analysis of HTLV-I proviral load in 202 HAM/TSP patients and 243 asymptomatic HTLV-I carriers: high proviral load strongly predisposes to HAM/TSP. J Neurovirol. 1998;4(6):586–593. - PubMed
-
- Etoh K, Yamaguchi K, Tokudome S, et al. Rapid quantification of HTLV-I provirus load: detection of monoclonal proliferation of HTLV-I-infected cells among blood donors. Int J Cancer. 1999;81(6):859–864. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
