

GiRaF: robust, computational identification of influenza reassortments via graph mining
- PMID: 21177643
- PMCID: PMC3064795
- DOI: 10.1093/nar/gkq1232
GiRaF: robust, computational identification of influenza reassortments via graph mining
Abstract
Reassortments in the influenza virus--a process where strains exchange genetic segments--have been implicated in two out of three pandemics of the 20th century as well as the 2009 H1N1 outbreak. While advances in sequencing have led to an explosion in the number of whole-genome sequences that are available, an understanding of the rate and distribution of reassortments and their role in viral evolution is still lacking. An important factor in this is the paucity of automated tools for confident identification of reassortments from sequence data due to the challenges of analyzing large, uncertain viral phylogenies. We describe here a novel computational method, called GiRaF (Graph-incompatibility-based Reassortment Finder), that robustly identifies reassortments in a fully automated fashion while accounting for uncertainties in the inferred phylogenies. The algorithms behind GiRaF search large collections of Markov chain Monte Carlo (MCMC)-sampled trees for groups of incompatible splits using a fast biclique enumeration algorithm coupled with several statistical tests to identify sets of taxa with differential phylogenetic placement. GiRaF correctly finds known reassortments in human, avian, and swine influenza populations, including the evolutionary events that led to the recent 'swine flu' outbreak. GiRaF also identifies several previously unreported reassortments via whole-genome studies to catalog events in H5N1 and swine influenza isolates.
Figures
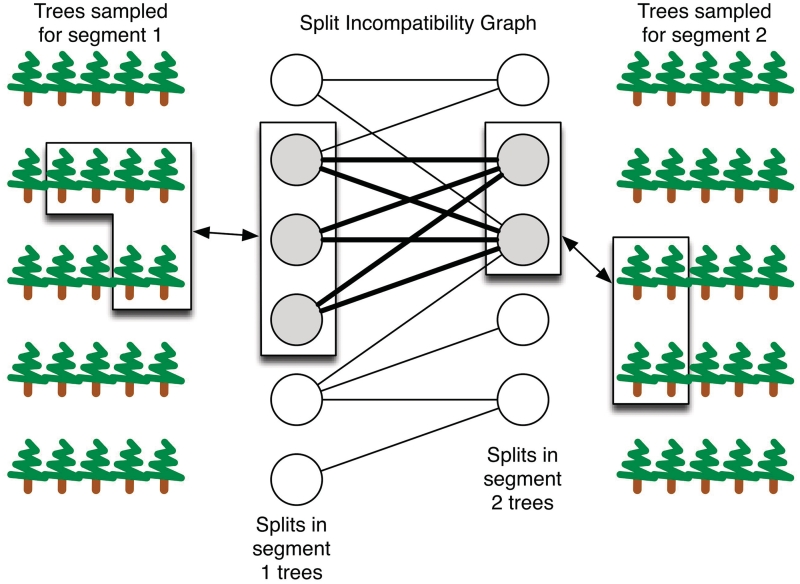
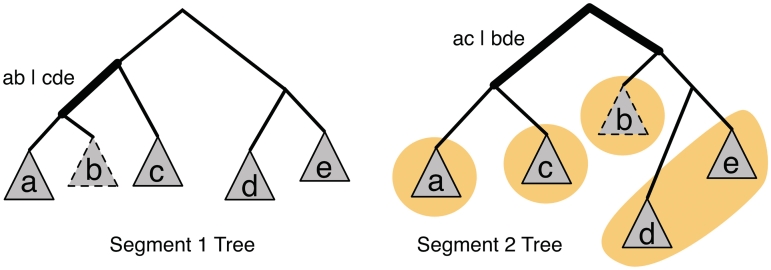
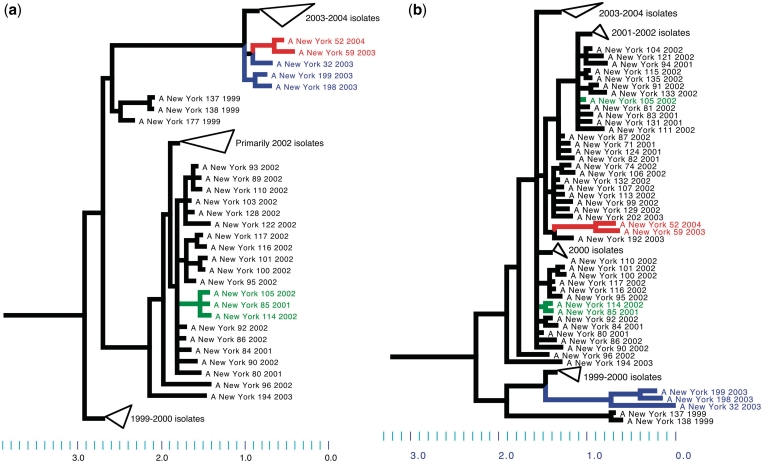
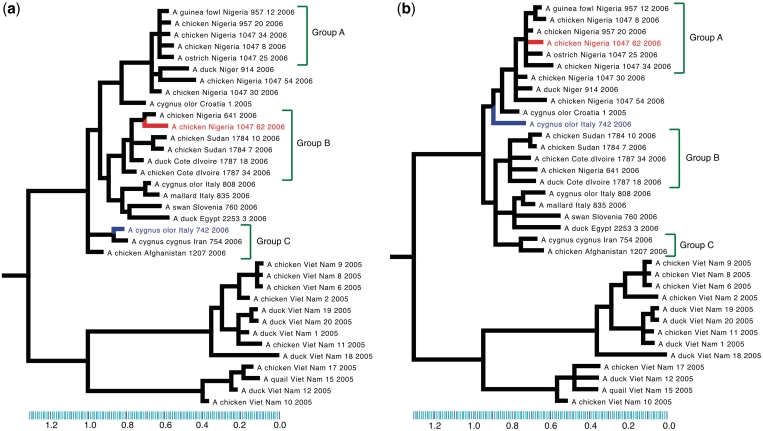
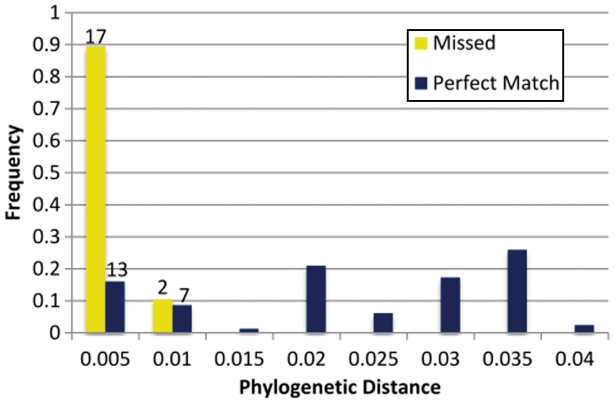
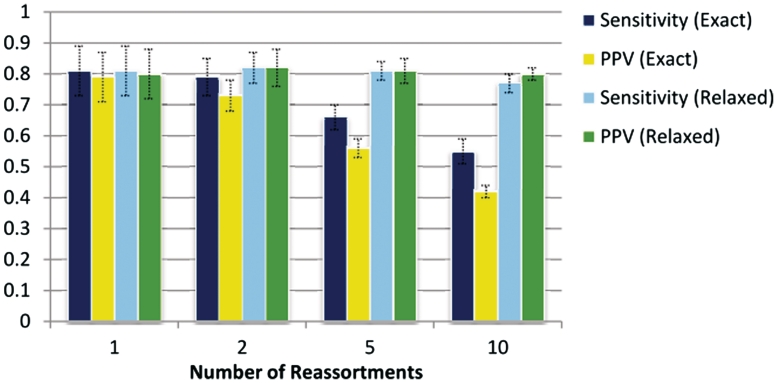
Similar articles
-
FluReF, an automated flu virus reassortment finder based on phylogenetic trees.BMC Genomics. 2011;12 Suppl 2(Suppl 2):S3. doi: 10.1186/1471-2164-12-S2-S3. Epub 2011 Jul 27. BMC Genomics. 2011. PMID: 21989112 Free PMC article.
-
Influenza A Viruses of Swine (IAV-S) in Vietnam from 2010 to 2015: Multiple Introductions of A(H1N1)pdm09 Viruses into the Pig Population and Diversifying Genetic Constellations of Enzootic IAV-S.J Virol. 2016 Dec 16;91(1):e01490-16. doi: 10.1128/JVI.01490-16. Print 2017 Jan 1. J Virol. 2016. PMID: 27795418 Free PMC article.
-
A phylogenetic approach to detecting reassortments in viruses with segmented genomes.Gene. 2010 Sep 15;464(1-2):11-6. doi: 10.1016/j.gene.2010.05.002. Epub 2010 May 28. Gene. 2010. PMID: 20546849
-
[Swine influenza virus: evolution mechanism and epidemic characterization--a review].Wei Sheng Wu Xue Bao. 2009 Sep;49(9):1138-45. Wei Sheng Wu Xue Bao. 2009. PMID: 20030049 Review. Chinese.
-
Progress and Challenge in Computational Identification of Influenza Virus Reassortment.Virol Sin. 2021 Dec;36(6):1273-1283. doi: 10.1007/s12250-021-00392-w. Epub 2021 May 26. Virol Sin. 2021. PMID: 34037948 Free PMC article. Review.
Cited by
-
Genetic evolution of the neuraminidase of influenza A (H3N2) viruses from 1968 to 2009 and its correspondence to haemagglutinin evolution.J Gen Virol. 2012 Sep;93(Pt 9):1996-2007. doi: 10.1099/vir.0.043059-0. Epub 2012 Jun 20. J Gen Virol. 2012. PMID: 22718569 Free PMC article.
-
Temporal and Gene Reassortment Analysis of Influenza C Virus Outbreaks in Hong Kong, SAR, China.J Virol. 2022 Feb 9;96(3):e0192821. doi: 10.1128/JVI.01928-21. Epub 2021 Nov 17. J Virol. 2022. PMID: 34787455 Free PMC article.
-
Unraveling the web of viroinformatics: computational tools and databases in virus research.J Virol. 2015 Feb;89(3):1489-501. doi: 10.1128/JVI.02027-14. Epub 2014 Nov 26. J Virol. 2015. PMID: 25428870 Free PMC article. Review.
-
Contrasting selective patterns across the segmented genome of bluetongue virus in a global reassortment hotspot.Virus Evol. 2019 Aug 5;5(2):vez027. doi: 10.1093/ve/vez027. eCollection 2019 Jul. Virus Evol. 2019. PMID: 31392031 Free PMC article.
-
Genome-informed investigation of the molecular evolution and genetic reassortment of severe fever with thrombocytopenia syndrome virus.PLoS Negl Trop Dis. 2023 Sep 15;17(9):e0011630. doi: 10.1371/journal.pntd.0011630. eCollection 2023 Sep. PLoS Negl Trop Dis. 2023. PMID: 37713429 Free PMC article.
References
-
- Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 2009;360:2605–2615. - PubMed
-
- Ghedin E, Sengamalay NA, Shumway M, Zaborsky J, Feldblyum T, Subbu V, Spiro DJ, Sitz J, Koo H, Bolotov P, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–1166. - PubMed
