

Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation
- PMID: 20388715
- PMCID: PMC2885238
- DOI: 10.1074/jbc.M109.069955
Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation
Abstract
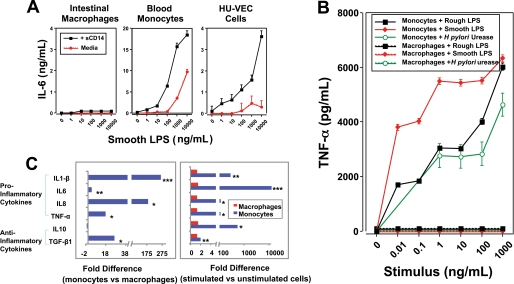
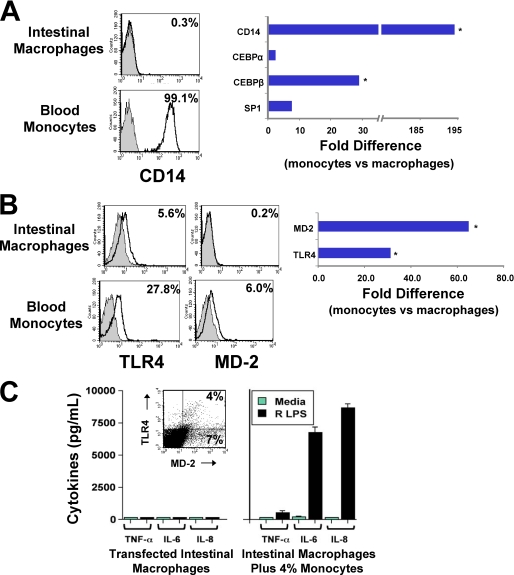
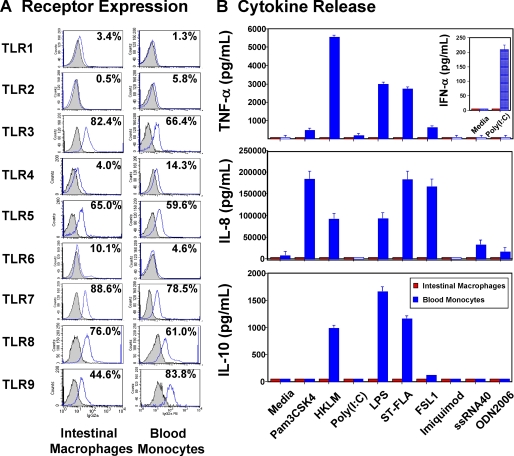
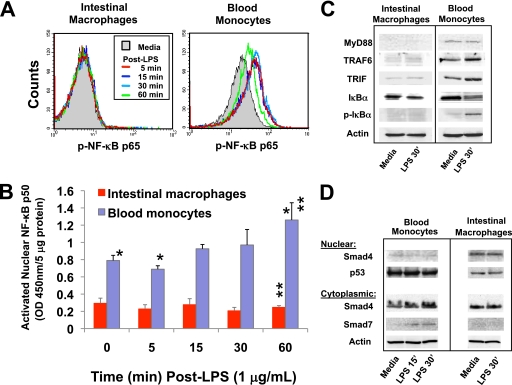
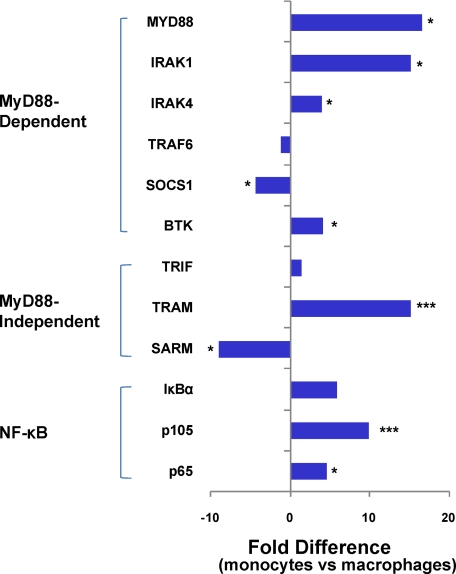
Human intestinal macrophages contribute to tissue homeostasis in noninflamed mucosa through profound down-regulation of pro-inflammatory cytokine release. Here, we show that this down-regulation extends to Toll-like receptor (TLR)-induced cytokine release, as intestinal macrophages expressed TLR3-TLR9 but did not release cytokines in response to TLR-specific ligands. Likely contributing to this unique functional profile, intestinal macrophages expressed markedly down-regulated adapter proteins MyD88 and Toll interleukin receptor 1 domain-containing adapter-inducing interferon beta, which together mediate all TLR MyD88-dependent and -independent NF-kappaB signaling, did not phosphorylate NF-kappaB p65 or Smad-induced IkappaBalpha, and did not translocate NF-kappaB into the nucleus. Importantly, transforming growth factor-beta released from intestinal extracellular matrix (stroma) induced identical down-regulation in the NF-kappaB signaling and function of blood monocytes, the exclusive source of intestinal macrophages. Our findings implicate stromal transforming growth factor-beta-induced dysregulation of NF-kappaB proteins and Smad signaling in the differentiation of pro-inflammatory blood monocytes into noninflammatory intestinal macrophages.
Figures

Similar articles
-
Intestinal macrophages and response to microbial encroachment.Mucosal Immunol. 2011 Jan;4(1):31-42. doi: 10.1038/mi.2010.66. Epub 2010 Oct 20. Mucosal Immunol. 2011. PMID: 20962772 Free PMC article. Review.
-
Oral administration of transforming growth factor-β1 (TGF-β1) protects the immature gut from injury via Smad protein-dependent suppression of epithelial nuclear factor κB (NF-κB) signaling and proinflammatory cytokine production.J Biol Chem. 2013 Nov 29;288(48):34757-66. doi: 10.1074/jbc.M113.503946. Epub 2013 Oct 15. J Biol Chem. 2013. PMID: 24129565 Free PMC article.
-
Transforming growth factor-beta -Smad signaling pathway cooperates with NF-kappa B to mediate nontypeable Haemophilus influenzae-induced MUC2 mucin transcription.J Biol Chem. 2002 Nov 22;277(47):45547-57. doi: 10.1074/jbc.M206883200. Epub 2002 Sep 16. J Biol Chem. 2002. PMID: 12237307
-
Cytomegalovirus promotes intestinal macrophage-mediated mucosal inflammation through induction of Smad7.Mucosal Immunol. 2018 Nov;11(6):1694-1704. doi: 10.1038/s41385-018-0041-4. Epub 2018 Aug 3. Mucosal Immunol. 2018. PMID: 30076393 Free PMC article.
-
Regulation of the MIR155 host gene in physiological and pathological processes.Gene. 2013 Dec 10;532(1):1-12. doi: 10.1016/j.gene.2012.12.009. Epub 2012 Dec 14. Gene. 2013. PMID: 23246696 Review.
Cited by
-
Differential susceptibility to lipopolysaccharide affects the activation of toll-like-receptor 4 signaling in THP-1 cells and PMA-differentiated THP-1 cells.Innate Immun. 2022 Apr;28(3-4):122-129. doi: 10.1177/17534259221100170. Innate Immun. 2022. PMID: 35612375 Free PMC article.
-
Exosomes in the gut.Front Immunol. 2014 Mar 17;5:104. doi: 10.3389/fimmu.2014.00104. eCollection 2014. Front Immunol. 2014. PMID: 24672525 Free PMC article. Review. No abstract available.
-
Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children.Mucosal Immunol. 2013 Sep;6(5):950-959. doi: 10.1038/mi.2012.133. Epub 2013 Jan 9. Mucosal Immunol. 2013. PMID: 23299619 Free PMC article.
-
Host-microbiota interactions in inflammatory bowel disease.Gut Microbes. 2012 Jul-Aug;3(4):332-44. doi: 10.4161/gmic.20228. Epub 2012 May 10. Gut Microbes. 2012. PMID: 22572873 Free PMC article. Review.
-
Overexpression of microRNA-21 is associated with elevated pro-inflammatory cytokines in dominant-negative TGF-β receptor type II mouse.J Autoimmun. 2013 Mar;41:111-9. doi: 10.1016/j.jaut.2012.12.013. Epub 2013 Feb 8. J Autoimmun. 2013. PMID: 23395552 Free PMC article.
References
-
- Smythies L. E., Maheshwari A., Clements R., Eckhoff D., Novak L., Vu H. L., Mosteller-Barnum L. M., Sellers M., Smith P. D. (2006) J. Leukocyte Biol. 80, 492–499 - PubMed
-
- Smith P. D., Smythies L. E., Mosteller-Barnum M., Sibley D. A., Russell M. W., Merger M., Sellers M. T., Orenstein J. M., Shimada T., Graham M. F., Kubagawa H. (2001) J. Immunol. 167, 2651–2656 - PubMed
-
- Smith P. D., Ochsenbauer-Jambor C., Smythies L. E. (2005) Immunol. Rev. 206, 149–159 - PubMed
-
- Smith P. D., Janoff E. N., Mosteller-Barnum M., Merger M., Orenstein J. M., Kearney J. F., Graham M. F. (1997) J. Immunol. Methods 202, 1–11 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DK-61297/DK/NIDDK NIH HHS/United States
- R21 AI083539/AI/NIAID NIH HHS/United States
- DK-47322/DK/NIDDK NIH HHS/United States
- R01 DK043183/DK/NIDDK NIH HHS/United States
- RR-20136/RR/NCRR NIH HHS/United States
- R01 DK047322/DK/NIDDK NIH HHS/United States
- DK-64400/DK/NIDDK NIH HHS/United States
- P01 AI083027/AI/NIAID NIH HHS/United States
- DK-54495/DK/NIDDK NIH HHS/United States
- R24 DK064400/DK/NIDDK NIH HHS/United States
- DK-74033/DK/NIDDK NIH HHS/United States
- C06 RR020136/RR/NCRR NIH HHS/United States
- DK-43183/DK/NIDDK NIH HHS/United States
- AI-83027/AI/NIAID NIH HHS/United States
- R21 DK074033/DK/NIDDK NIH HHS/United States
- R01 DK054495/DK/NIDDK NIH HHS/United States
- AI-83539/AI/NIAID NIH HHS/United States
- I01 CX000372/CX/CSRD VA/United States
- R01 DK061297/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
