

The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer
- PMID: 20385749
- PMCID: PMC2867287
- DOI: 10.1084/jem.20100050
The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer
Abstract
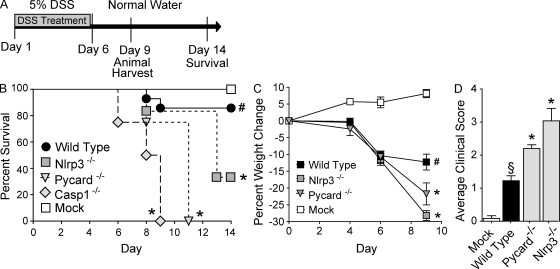
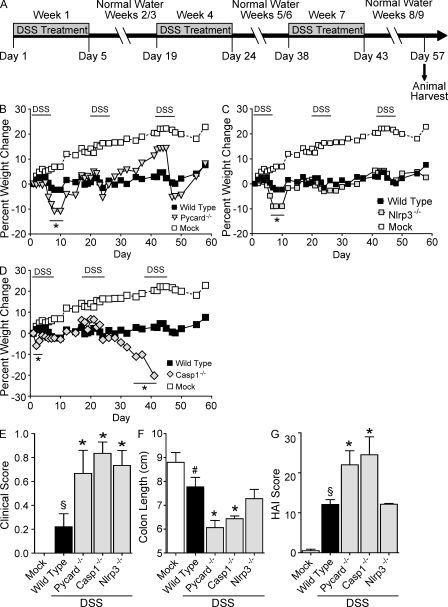
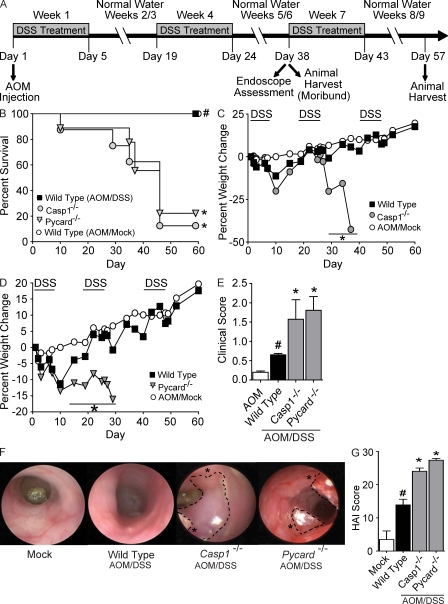
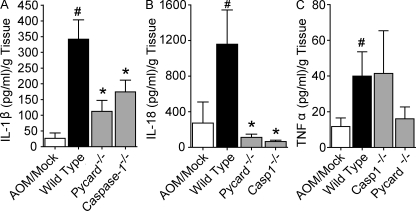
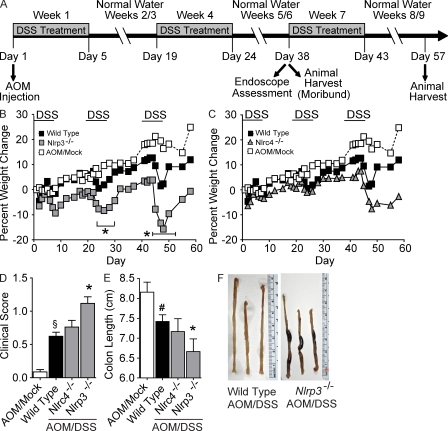
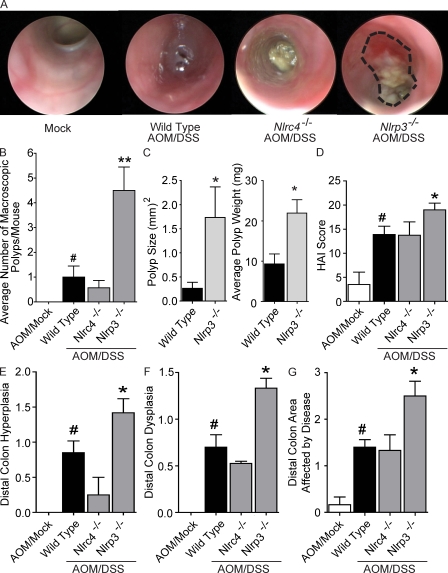
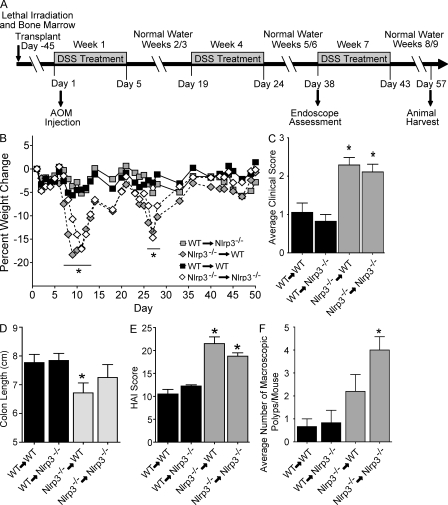
Colitis-associated cancer (CAC) is a major complication of inflammatory bowel diseases. We show that components of the inflammasome are protective during acute and recurring colitis and CAC in the dextran sulfate sodium (DSS) and azoxymethane + DSS models. Mice lacking the inflammasome adaptor protein PYCARD (ASC) and caspase-1 demonstrate increased disease outcome, morbidity, histopathology, and polyp formation. The increased tumor burden is correlated with attenuated levels of IL-1beta and IL-18 at the tumor site. To decipher the nucleotide-binding domain, leucine-rich-repeat-containing (NLR) component that is involved in colitis and CAC, we assessed Nlrp3 and Nlrc4 deficient mice. Nlrp3(-/-) mice showed an increase in acute and recurring colitis and CAC, although the disease outcome was less severe in Nlrp3(-/-) mice than in Pycard(-/-) or Casp1(-/-) animals. No significant differences were observed in disease progression or outcome in Nlrc4(-/-) mice compared with similarly treated wild-type animals. Bone marrow reconstitution experiments show that Nlrp3 gene expression and function in hematopoietic cells, rather than intestinal epithelial cells or stromal cells, is responsible for protection against increased tumorigenesis. These data suggest that the inflammasome functions as an attenuator of colitis and CAC.
Figures
Comment in
-
Immunology: The inflammasome protects?Nat Rev Cancer. 2010 Jun;10(6):383. doi: 10.1038/nrc2862. Nat Rev Cancer. 2010. PMID: 20506588 No abstract available.
Similar articles
-
Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer.Autophagy. 2014 Jun;10(6):972-85. doi: 10.4161/auto.28374. Autophagy. 2014. PMID: 24879148 Free PMC article.
-
The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis.Immunity. 2010 Mar 26;32(3):379-91. doi: 10.1016/j.immuni.2010.03.003. Epub 2010 Mar 18. Immunity. 2010. PMID: 20303296 Free PMC article.
-
Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome.Gut. 2010 Sep;59(9):1192-9. doi: 10.1136/gut.2009.197822. Epub 2010 May 4. Gut. 2010. PMID: 20442201
-
[Inflammatory bowel diseases and inflammasome].Korean J Gastroenterol. 2011 Dec;58(6):300-10. doi: 10.4166/kjg.2011.58.6.300. Korean J Gastroenterol. 2011. PMID: 22198227 Review. Korean.
-
The Nlrp3 inflammasome: contributions to intestinal homeostasis.Trends Immunol. 2011 Apr;32(4):171-9. doi: 10.1016/j.it.2011.02.002. Trends Immunol. 2011. PMID: 21388882 Free PMC article. Review.
Cited by
-
Targeting ROS in cancer: rationale and strategies.Nat Rev Drug Discov. 2024 Aug;23(8):583-606. doi: 10.1038/s41573-024-00979-4. Epub 2024 Jul 9. Nat Rev Drug Discov. 2024. PMID: 38982305 Review.
-
Compound loss of GSDMD and GSDME function is necessary to achieve maximal therapeutic effect in colitis.J Transl Autoimmun. 2022 Aug 30;5:100162. doi: 10.1016/j.jtauto.2022.100162. eCollection 2022. J Transl Autoimmun. 2022. PMID: 36097634 Free PMC article.
-
Microbiota as a mediator of cancer progression and therapy.Transl Res. 2017 Jan;179:139-154. doi: 10.1016/j.trsl.2016.07.021. Epub 2016 Aug 3. Transl Res. 2017. PMID: 27554797 Free PMC article. Review.
-
p16INK4a Plays Critical Role in Exacerbating Inflammaging in High Fat Diet Induced Skin.Oxid Med Cell Longev. 2022 Nov 21;2022:3415528. doi: 10.1155/2022/3415528. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 36457728 Free PMC article.
-
Lack of reelin modifies the gene expression in the small intestine of mice.J Physiol Biochem. 2012 Jun;68(2):205-18. doi: 10.1007/s13105-011-0132-0. Epub 2011 Dec 8. J Physiol Biochem. 2012. PMID: 22161684
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK073338/DK/NIDDK NIH HHS/United States
- F32 AI082895/AI/NIAID NIH HHS/United States
- P30 DK034987/DK/NIDDK NIH HHS/United States
- T32 HD046369/HD/NICHD NIH HHS/United States
- U19-AI077437-01/AI/NIAID NIH HHS/United States
- P30DK34987/DK/NIDDK NIH HHS/United States
- T32HD046369/HD/NICHD NIH HHS/United States
- T32 CA009156/CA/NCI NIH HHS/United States
- T32CA009156/CA/NCI NIH HHS/United States
- T32 AR007416/AR/NIAMS NIH HHS/United States
- U19 AI077437/AI/NIAID NIH HHS/United States
- F32AI082895/AI/NIAID NIH HHS/United States
- T32AR007416/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
