

Insulin signaling regulates mitochondrial function in pancreatic beta-cells
- PMID: 19956695
- PMCID: PMC2776992
- DOI: 10.1371/journal.pone.0007983
Insulin signaling regulates mitochondrial function in pancreatic beta-cells
Abstract
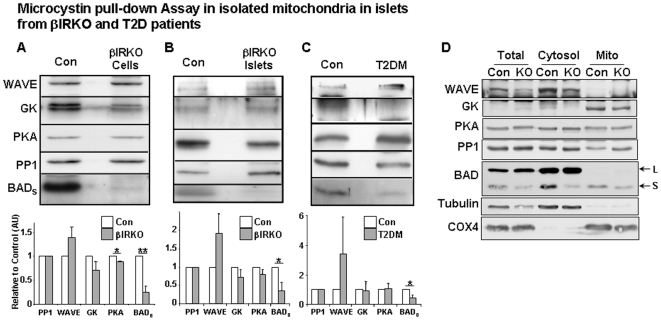
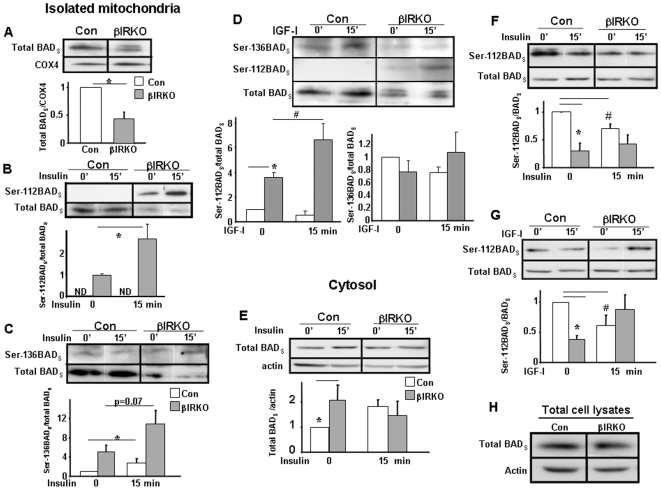
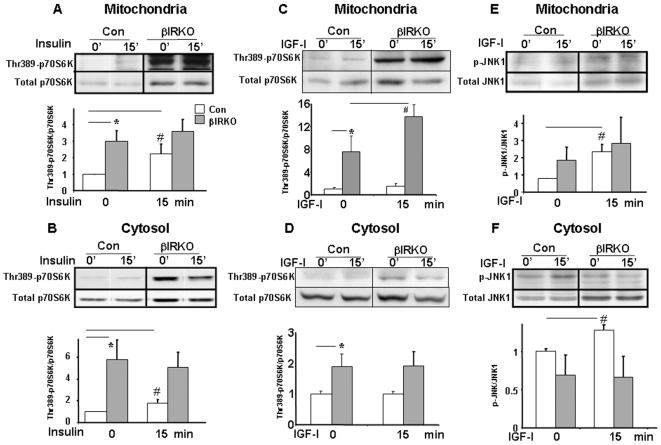
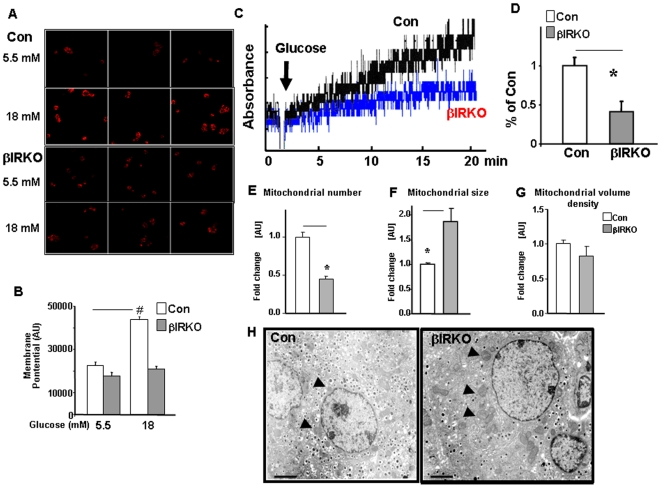
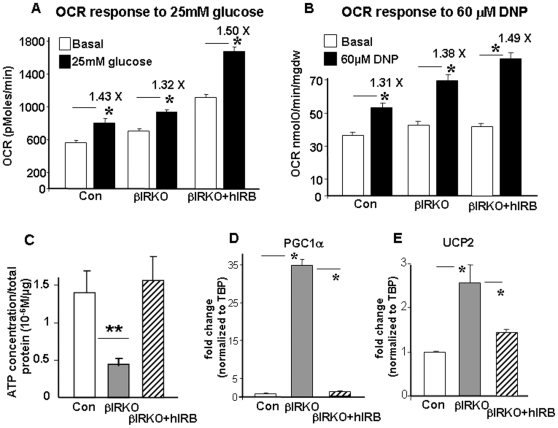
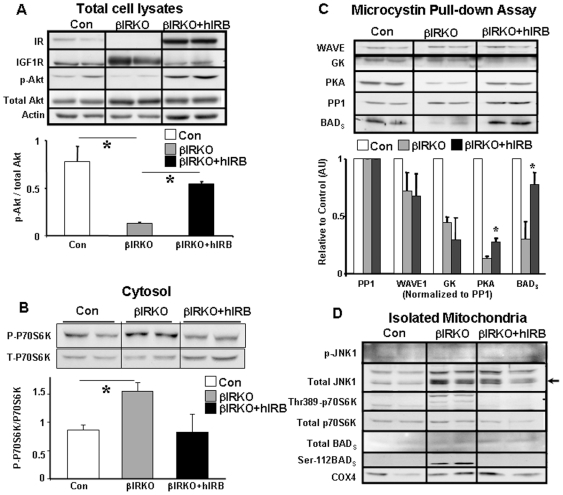
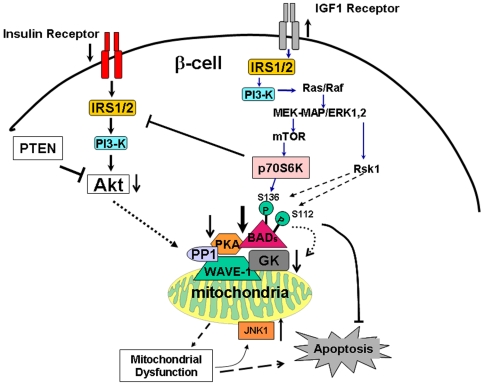
Insulin/IGF-I signaling regulates the metabolism of most mammalian tissues including pancreatic islets. To dissect the mechanisms linking insulin signaling with mitochondrial function, we first identified a mitochondria-tethering complex in beta-cells that included glucokinase (GK), and the pro-apoptotic protein, BAD(S). Mitochondria isolated from beta-cells derived from beta-cell specific insulin receptor knockout (betaIRKO) mice exhibited reduced BAD(S), GK and protein kinase A in the complex, and attenuated function. Similar alterations were evident in islets from patients with type 2 diabetes. Decreased mitochondrial GK activity in betaIRKOs could be explained, in part, by reduced expression and altered phosphorylation of BAD(S). The elevated phosphorylation of p70S6K and JNK1 was likely due to compensatory increase in IGF-1 receptor expression. Re-expression of insulin receptors in betaIRKO cells partially restored the stoichiometry of the complex and mitochondrial function. These data indicate that insulin signaling regulates mitochondrial function and have implications for beta-cell dysfunction in type 2 diabetes.
Conflict of interest statement
Figures
Similar articles
-
Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus.PLoS One. 2011;6(11):e28050. doi: 10.1371/journal.pone.0028050. Epub 2011 Nov 30. PLoS One. 2011. PMID: 22140505 Free PMC article.
-
Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice.Am J Physiol Endocrinol Metab. 2004 Jan;286(1):E41-9. doi: 10.1152/ajpendo.00533.2001. Epub 2003 Sep 30. Am J Physiol Endocrinol Metab. 2004. PMID: 14519599
-
Octanoic acid potentiates glucose-stimulated insulin secretion and expression of glucokinase through the olfactory receptor in pancreatic β-cells.Biochem Biophys Res Commun. 2018 Sep 3;503(1):278-284. doi: 10.1016/j.bbrc.2018.06.015. Epub 2018 Jun 11. Biochem Biophys Res Commun. 2018. PMID: 29885841
-
Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm.Diabetes. 1996 Feb;45(2):223-41. doi: 10.2337/diab.45.2.223. Diabetes. 1996. PMID: 8549869 Review.
-
Modulation of β-cell function: a translational journey from the bench to the bedside.Diabetes Obes Metab. 2012 Oct;14 Suppl 3:152-60. doi: 10.1111/j.1463-1326.2012.01647.x. Diabetes Obes Metab. 2012. PMID: 22928576 Review.
Cited by
-
Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus.PLoS One. 2011;6(11):e28050. doi: 10.1371/journal.pone.0028050. Epub 2011 Nov 30. PLoS One. 2011. PMID: 22140505 Free PMC article.
-
Insulin signaling meets mitochondria in metabolism.Trends Endocrinol Metab. 2010 Oct;21(10):589-98. doi: 10.1016/j.tem.2010.06.005. Epub 2010 Jul 16. Trends Endocrinol Metab. 2010. PMID: 20638297 Free PMC article. Review.
-
Basal alpha-cell up-regulation in obese insulin-resistant adolescents.J Clin Endocrinol Metab. 2011 Jan;96(1):91-7. doi: 10.1210/jc.2010-1275. Epub 2010 Sep 15. J Clin Endocrinol Metab. 2011. PMID: 20843946 Free PMC article.
-
Insulin infusion during normoglycemia modulates insulin secretion according to whole-body insulin sensitivity.Diabetes Care. 2011 Feb;34(2):437-41. doi: 10.2337/dc10-1137. Epub 2011 Jan 7. Diabetes Care. 2011. PMID: 21216852 Free PMC article. Clinical Trial.
-
Brain energy metabolism parameters in an animal model of diabetes.Metab Brain Dis. 2010 Dec;25(4):391-6. doi: 10.1007/s11011-010-9220-z. Epub 2010 Nov 19. Metab Brain Dis. 2010. PMID: 21088877
References
-
- Kim JJ, Accili D. Signalling through IGF-I and insulin receptors: where is the specificity? Growth Horm IGF Res. 2002;12:84–90. - PubMed
-
- Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. - PubMed
-
- Otani K, Kulkarni RN, Baldwin AC, Krutzfeldt J, Ueki K, et al. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am J Physiol Endocrinol Metab. 2004;286:E41–49. - PubMed
-
- Kulkarni RN, Holzenberger M, Shih DQ, Ozcan U, Stoffel M, et al. beta-cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter beta-cell mass. Nat Genet. 2002;31:111–115. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
