

A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP
- PMID: 19675240
- PMCID: PMC2749997
- DOI: 10.1523/JNEUROSCI.2542-09.2009
A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP
Abstract
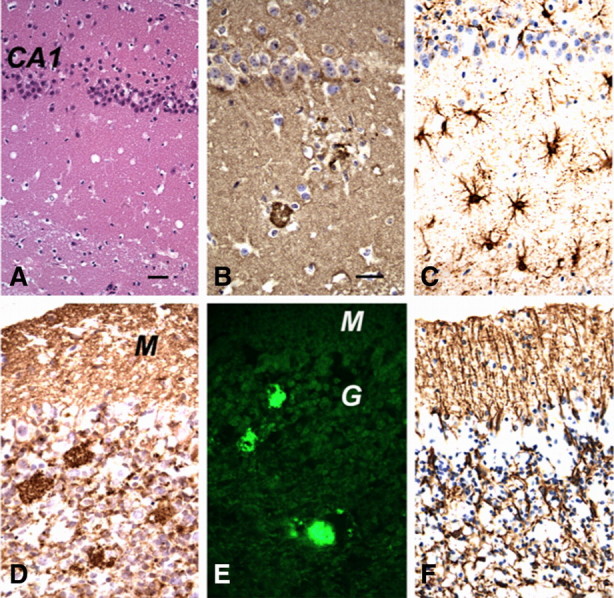
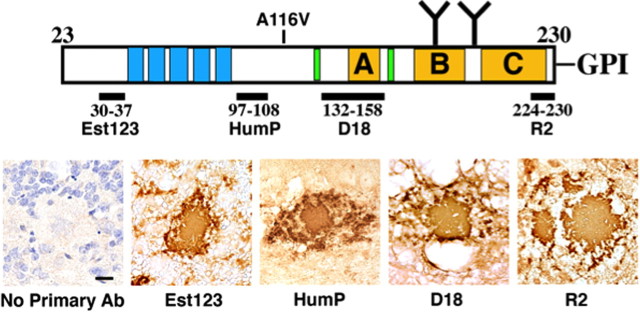
Gerstmann-Sträussler-Scheinker syndrome (GSS) is a genetic prion disease typified clinically by the development of progressive ataxia and dementia, and histopathologically by the presence of prion protein (PrP) amyloid plaques in the CNS, especially within the cerebellum. Several mutations of the PrP gene (PRNP) are associated with GSS, but only the P102L mutation has been convincingly modeled in transgenic (Tg) mice. To determine whether other mutations carry specific GSS phenotypic information, we constructed Tg mice that express PrP carrying the mouse homolog of the GSS-associated A117V mutation. Tg(A116V) mice express approximately six times the endogenous levels of PrP, develop progressive ataxia by approximately 140 d, and die by approximately 170 d. Compared with a mouse model of transmissible Creutzfeldt-Jakob disease (CJD), the ataxia of Tg(A116V) mice is more prominent, and the course of disease is more protracted, paralleling that observed in human disease. Neuropathology includes mild scattered vacuolation and prominent, mainly cerebellar localized, thioflavin S-positive PrP plaques comprised of full-length PrP(A116V). In some mice, more prominent vacuolation or a noncerebellar distribution of PrP plaques was evident, suggesting some variability in phenotype. The biophysical properties of PrP from Tg(A116V) mice and human GSS(A117V) revealed a similarly low fraction of insoluble PrP and a weakly protease-resistant approximately 13 kDa midspan PrP fragment, not observed in CJD. Overall, Tg(A116V) mice recapitulate many clinicopathologic features of GSS(A117V) that are distinct from CJD, supporting PrP(A116V) to carry specific phenotypic information. The occasional variation in histopathology they exhibit may shed light on a similar observation in human GSS(A117V).
Figures






Similar articles
-
Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein.PLoS Pathog. 2013;9(9):e1003643. doi: 10.1371/journal.ppat.1003643. Epub 2013 Sep 26. PLoS Pathog. 2013. PMID: 24086135 Free PMC article.
-
Towards authentic transgenic mouse models of heritable PrP prion diseases.Acta Neuropathol. 2016 Oct;132(4):593-610. doi: 10.1007/s00401-016-1585-6. Epub 2016 Jun 28. Acta Neuropathol. 2016. PMID: 27350609 Free PMC article.
-
Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity.J Neuropathol Exp Neurol. 1998 Oct;57(10):979-88. doi: 10.1097/00005072-199810000-00010. J Neuropathol Exp Neurol. 1998. PMID: 9786248
-
Transmissibility of Gerstmann-Sträussler-Scheinker syndrome in rodent models: New insights into the molecular underpinnings of prion infectivity.Prion. 2016 Nov;10(6):421-433. doi: 10.1080/19336896.2016.1239686. Prion. 2016. PMID: 27892798 Free PMC article. Review.
-
Genetic and infectious prion diseases.Arch Neurol. 1993 Nov;50(11):1129-53. doi: 10.1001/archneur.1993.00540110011002. Arch Neurol. 1993. PMID: 8105771 Review.
Cited by
-
Therapeutic Trial of anle138b in Mouse Models of Genetic Prion Disease.J Virol. 2023 Feb 28;97(2):e0167222. doi: 10.1128/jvi.01672-22. Epub 2023 Jan 18. J Virol. 2023. PMID: 36651748 Free PMC article.
-
Bona fide atypical scrapie faithfully reproduced for the first time in a rodent model.Acta Neuropathol Commun. 2022 Dec 13;10(1):179. doi: 10.1186/s40478-022-01477-7. Acta Neuropathol Commun. 2022. PMID: 36514160 Free PMC article.
-
Mouse models for studying the formation and propagation of prions.J Biol Chem. 2014 Jul 18;289(29):19841-9. doi: 10.1074/jbc.R114.550707. Epub 2014 May 23. J Biol Chem. 2014. PMID: 24860095 Free PMC article. Review.
-
Generation of a new infectious recombinant prion: a model to understand Gerstmann-Sträussler-Scheinker syndrome.Sci Rep. 2017 Aug 29;7(1):9584. doi: 10.1038/s41598-017-09489-3. Sci Rep. 2017. PMID: 28851967 Free PMC article.
-
Convergent generation of atypical prions in knockin mouse models of genetic prion disease.J Clin Invest. 2024 Aug 1;134(15):e176344. doi: 10.1172/JCI176344. J Clin Invest. 2024. PMID: 39087478 Free PMC article.
References
-
- Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–529. - PubMed
-
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. - PubMed
-
- Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–19180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous