

An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis
- PMID: 19576851
- PMCID: PMC4718191
- DOI: 10.1016/S1470-2045(09)70164-0
An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis
Abstract
Background: Phaeochromocytomas and paragangliomas are neuro-endocrine tumours that occur sporadically and in several hereditary tumour syndromes, including the phaeochromocytoma-paraganglioma syndrome. This syndrome is caused by germline mutations in succinate dehydrogenase B (SDHB), C (SDHC), or D (SDHD) genes. Clinically, the phaeochromocytoma-paraganglioma syndrome is often unrecognised, although 10-30% of apparently sporadic phaeochromocytomas and paragangliomas harbour germline SDH-gene mutations. Despite these figures, the screening of phaeochromocytomas and paragangliomas for mutations in the SDH genes to detect phaeochromocytoma-paraganglioma syndrome is rarely done because of time and financial constraints. We investigated whether SDHB immunohistochemistry could effectively discriminate between SDH-related and non-SDH-related phaeochromocytomas and paragangliomas in large retrospective and prospective tumour series.
Methods: Immunohistochemistry for SDHB was done on 220 tumours. Two retrospective series of 175 phaeochromocytomas and paragangliomas with known germline mutation status for phaeochromocytoma-susceptibility or paraganglioma-susceptibility genes were investigated. Additionally, a prospective series of 45 phaeochromocytomas and paragangliomas was investigated for SDHB immunostaining followed by SDHB, SDHC, and SDHD mutation testing.
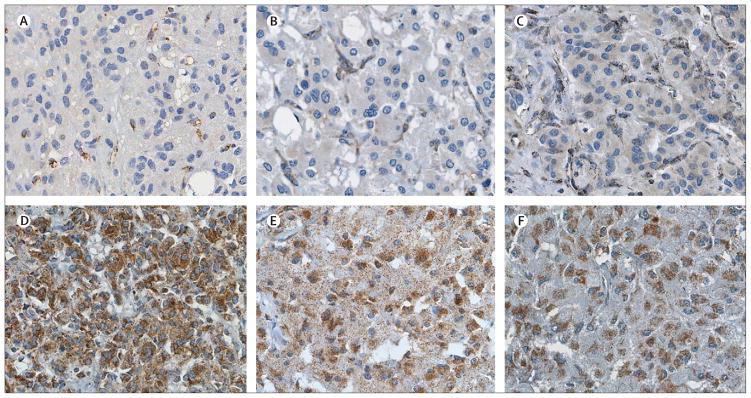
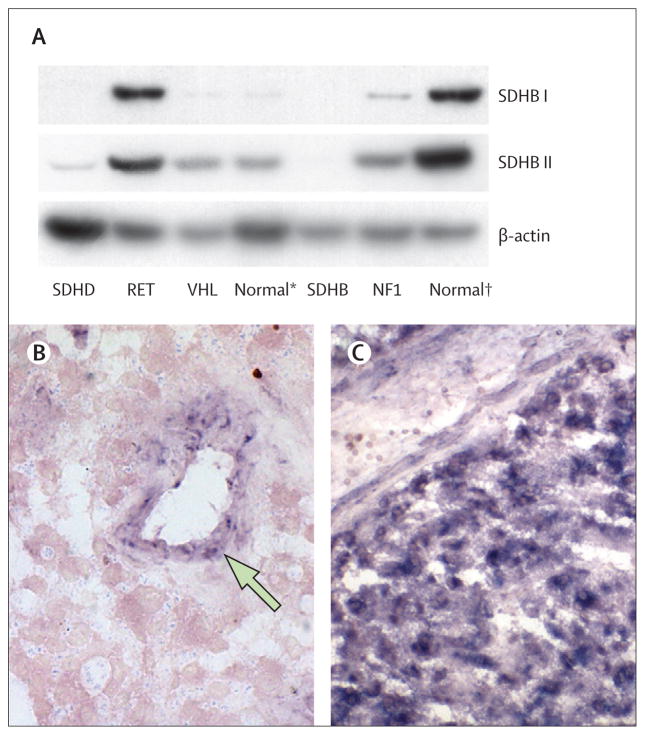
Findings: SDHB protein expression was absent in all 102 phaeochromocytomas and paragangliomas with an SDHB, SDHC, or SDHD mutation, but was present in all 65 paraganglionic tumours related to multiple endocrine neoplasia type 2, von Hippel-Lindau disease, and neurofibromatosis type 1. 47 (89%) of the 53 phaeochromocytomas and paragangliomas with no syndromic germline mutation showed SDHB expression. The sensitivity and specificity of the SDHB immunohistochemistry to detect the presence of an SDH mutation in the prospective series were 100% (95% CI 87-100) and 84% (60-97), respectively.
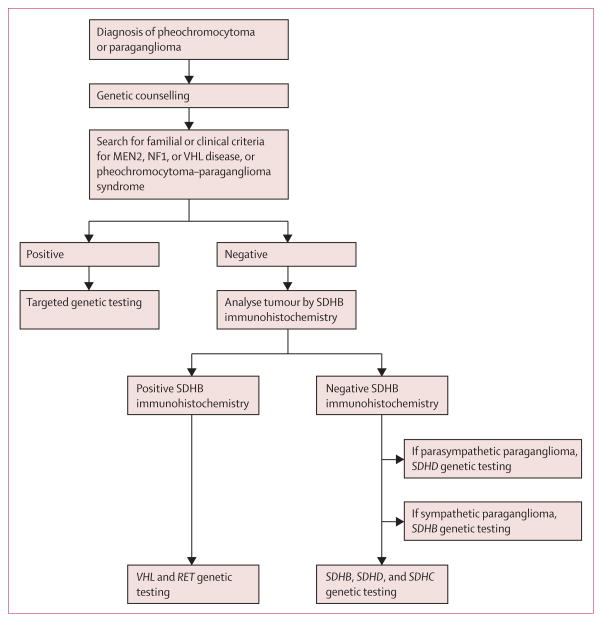
Interpretation: Phaeochromocytoma-paraganglioma syndrome can be diagnosed reliably by an immunohistochemical procedure. SDHB, SDHC, and SDHD germline mutation testing is indicated only in patients with SDHB-negative tumours. SDHB immunohistochemistry on phaeochromocytomas and paragangliomas could improve the diagnosis of phaeochromocytoma-paraganglioma syndrome.
Funding: The Netherlands Organisation for Scientific Research, Dutch Cancer Society, Vanderes Foundation, Association pour la Recherche contre le Cancer, Institut National de la Santé et de la Recherche Médicale, and a PHRC grant COMETE 3 for the COMETE network.
Conflict of interest statement
The authors declared no conflicts of interest.
Figures
Comment in
-
Diagnosing patients with hereditary paraganglial tumours.Lancet Oncol. 2009 Aug;10(8):741. doi: 10.1016/S1470-2045(09)70204-9. Lancet Oncol. 2009. PMID: 19647193 Review. No abstract available.
Similar articles
-
Potential Pitfalls of SDH Immunohistochemical Detection in Paragangliomas and Phaeochromocytomas Harbouring Germline SDHx Gene Mutation.Anticancer Res. 2017 Feb;37(2):805-812. doi: 10.21873/anticanres.11381. Anticancer Res. 2017. PMID: 28179334
-
Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas.Oncogene. 2003 Mar 6;22(9):1358-64. doi: 10.1038/sj.onc.1206300. Oncogene. 2003. PMID: 12618761
-
SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes.J Intern Med. 2009 Jul;266(1):19-42. doi: 10.1111/j.1365-2796.2009.02111.x. J Intern Med. 2009. PMID: 19522823 Free PMC article. Review.
-
Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD.J Med Genet. 2018 Jun;55(6):384-394. doi: 10.1136/jmedgenet-2017-105127. Epub 2018 Jan 31. J Med Genet. 2018. PMID: 29386252 Free PMC article.
-
Familial paraganglioma syndromes.J Clin Pathol. 2010 Jun;63(6):488-91. doi: 10.1136/jcp.2010.076257. J Clin Pathol. 2010. PMID: 20498024 Review.
Cited by
-
Metastatic Paraganglioma of the Spine With SDHB Mutation: Case Report and Review of the Literature.Int J Spine Surg. 2021 Feb;14(s4):S37-S45. doi: 10.14444/7163. Epub 2021 Jan 20. Int J Spine Surg. 2021. PMID: 33900943 Free PMC article.
-
Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing.Ann Surg Oncol. 2013 May;20(5):1444-50. doi: 10.1245/s10434-013-2942-5. Epub 2013 Mar 20. Ann Surg Oncol. 2013. PMID: 23512077 Free PMC article.
-
Prevalence of germline mutations in patients with pheochromocytoma or abdominal paraganglioma and sporadic presentation: a population-based study in Western Sweden.World J Surg. 2012 Jun;36(6):1389-94. doi: 10.1007/s00268-012-1430-6. World J Surg. 2012. PMID: 22270996 Free PMC article.
-
The Role of Immunohistochemistry and Molecular Analysis of Succinate Dehydrogenase in the Diagnosis of Endocrine and Non-Endocrine Tumors and Related Syndromes.Endocr Pathol. 2019 Mar;30(1):64-73. doi: 10.1007/s12022-018-9555-2. Endocr Pathol. 2019. PMID: 30421319 Review.
-
Overview of the 2022 WHO Classification of Familial Endocrine Tumor Syndromes.Endocr Pathol. 2022 Mar;33(1):197-227. doi: 10.1007/s12022-022-09705-5. Epub 2022 Mar 13. Endocr Pathol. 2022. PMID: 35285003 Review.
References
-
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665–75. - PubMed
-
- Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Phaeochromocytoma: an update on genetics and management. Endocr Relat Cancer. 2007;14:935–56. - PubMed
-
- Nakamura E, Kaelin WG., Jr Recent insights into the molecular pathogenesis of phaeochromocytoma and paraganglioma. Endocr Pathol. 2006;17:97–106. - PubMed
-
- Amar L, Bertherat J, Baudin E, et al. Genetic testing in phaeochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–18. - PubMed
-
- Lancaster CR. Succinate:quinone oxidoreductases: an overview. Biochim Biophys Acta. 2002;1553:1–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
