

The decapping activator Lsm1p-7p-Pat1p complex has the intrinsic ability to distinguish between oligoadenylated and polyadenylated RNAs
- PMID: 17513695
- PMCID: PMC1894922
- DOI: 10.1261/rna.502507
The decapping activator Lsm1p-7p-Pat1p complex has the intrinsic ability to distinguish between oligoadenylated and polyadenylated RNAs
Abstract
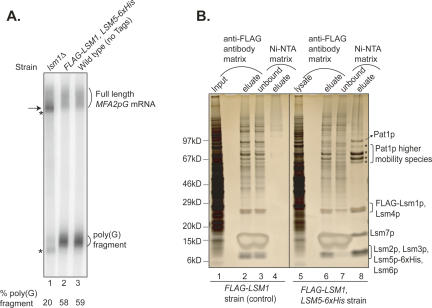
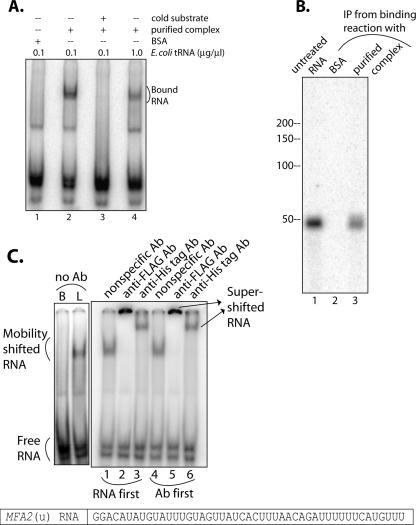
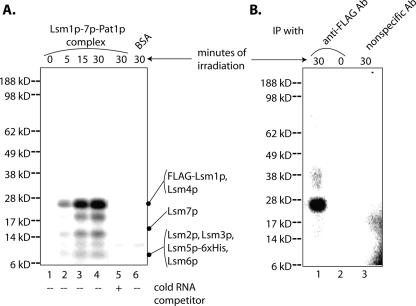
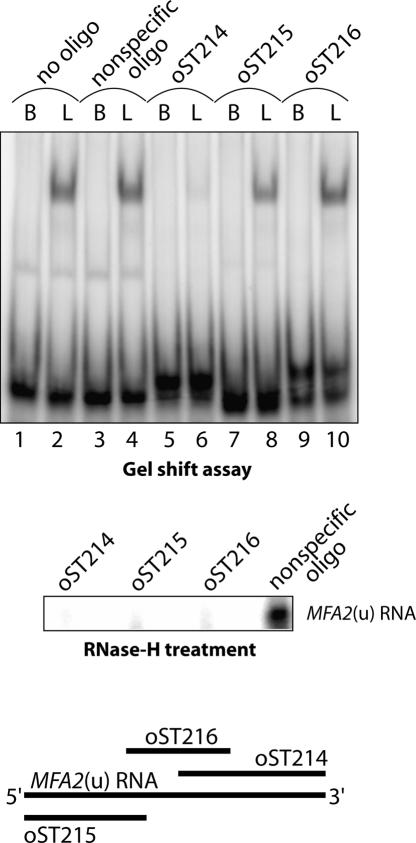
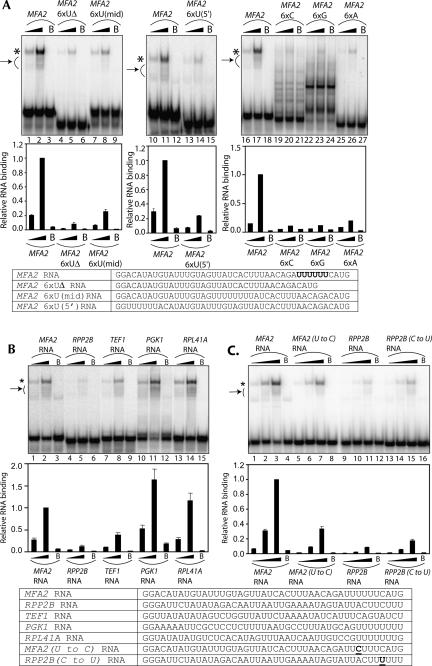
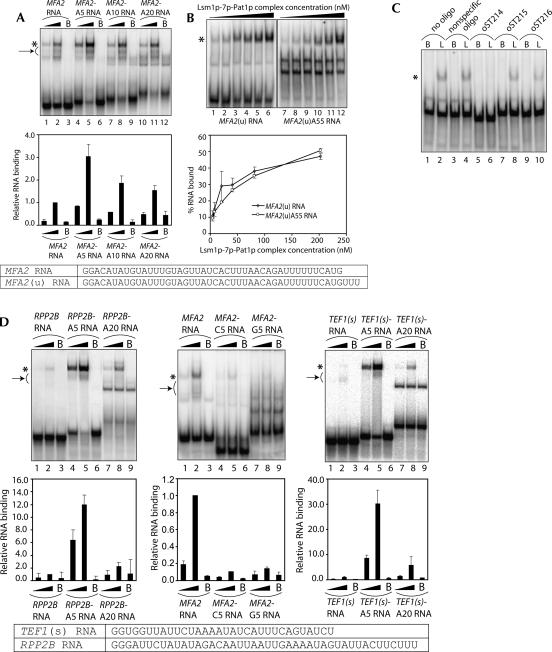
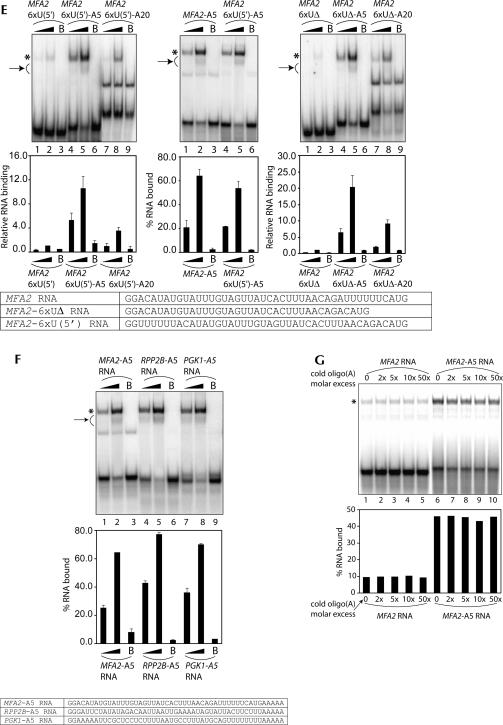
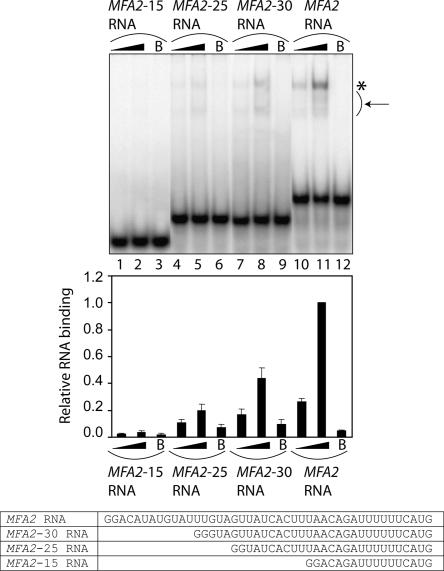
Decapping is a critical step in mRNA decay. In the 5'-to-3' mRNA decay pathway conserved in all eukaryotes, decay is initiated by poly(A) shortening, and oligoadenylated mRNAs (but not polyadenylated mRNAs) are selectively decapped allowing their subsequent degradation by 5' to 3' exonucleolysis. The highly conserved heptameric Lsm1p-7p complex (made up of the seven Sm-like proteins, Lsm1p-Lsm7p) and its interacting partner Pat1p activate decapping by an unknown mechanism and localize with other decapping factors to the P-bodies in the cytoplasm. The Lsm1p-7p-Pat1p complex also protects the 3'-ends of mRNAs in vivo from trimming, presumably by binding to the 3'-ends. In order to determine the intrinsic RNA-binding properties of this complex, we have purified it from yeast and carried out in vitro analyses. Our studies revealed that it directly binds RNA at/near the 3'-end. Importantly, it possesses the intrinsic ability to distinguish between oligoadenylated and polyadenylated RNAs such that the former are bound with much higher affinity than the latter. These results indicate that the intrinsic RNA-binding characteristics of this complex form a critical determinant of its in vivo interactions and functions.
Figures
Similar articles
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
Comparison of Two Modern Survival Prediction Tools, SORG-MLA and METSSS, in Patients With Symptomatic Long-bone Metastases Who Underwent Local Treatment With Surgery Followed by Radiotherapy and With Radiotherapy Alone.Clin Orthop Relat Res. 2024 Dec 1;482(12):2193-2208. doi: 10.1097/CORR.0000000000003185. Epub 2024 Jul 23. Clin Orthop Relat Res. 2024. PMID: 39051924
-
Qualitative evidence synthesis informing our understanding of people's perceptions and experiences of targeted digital communication.Cochrane Database Syst Rev. 2019 Oct 23;10(10):ED000141. doi: 10.1002/14651858.ED000141. Cochrane Database Syst Rev. 2019. PMID: 31643081 Free PMC article.
-
"I've Spent My Whole Life Striving to Be Normal": Internalized Stigma and Perceived Impact of Diagnosis in Autistic Adults.Autism Adulthood. 2023 Dec 1;5(4):423-436. doi: 10.1089/aut.2022.0066. Epub 2023 Dec 12. Autism Adulthood. 2023. PMID: 38116050 Free PMC article.
-
Pharmacological treatments in panic disorder in adults: a network meta-analysis.Cochrane Database Syst Rev. 2023 Nov 28;11(11):CD012729. doi: 10.1002/14651858.CD012729.pub3. Cochrane Database Syst Rev. 2023. PMID: 38014714 Free PMC article. Review.
Cited by
-
Beyond transcription factors: roles of mRNA decay in regulating gene expression in plants.F1000Res. 2018 Dec 17;7:F1000 Faculty Rev-1940. doi: 10.12688/f1000research.16203.1. eCollection 2018. F1000Res. 2018. PMID: 30613385 Free PMC article. Review.
-
RNA uridylation and decay in plants.Philos Trans R Soc Lond B Biol Sci. 2018 Nov 5;373(1762):20180163. doi: 10.1098/rstb.2018.0163. Philos Trans R Soc Lond B Biol Sci. 2018. PMID: 30397100 Free PMC article. Review.
-
Activation of decapping involves binding of the mRNA and facilitation of the post-binding steps by the Lsm1-7-Pat1 complex.RNA. 2009 Oct;15(10):1837-48. doi: 10.1261/rna.1650109. Epub 2009 Jul 30. RNA. 2009. PMID: 19643916 Free PMC article.
-
The ApaH-like phosphatase TbALPH1 is the major mRNA decapping enzyme of trypanosomes.PLoS Pathog. 2017 Jun 19;13(6):e1006456. doi: 10.1371/journal.ppat.1006456. eCollection 2017 Jun. PLoS Pathog. 2017. PMID: 28628654 Free PMC article.
-
Emerging roles for ribonucleoprotein modification and remodeling in controlling RNA fate.Trends Cell Biol. 2013 Oct;23(10):504-10. doi: 10.1016/j.tcb.2013.05.001. Epub 2013 Jun 4. Trends Cell Biol. 2013. PMID: 23756094 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases