

Utilizing the activation mechanism of serine proteases to engineer hepatocyte growth factor into a Met antagonist
- PMID: 17372204
- PMCID: PMC1828710
- DOI: 10.1073/pnas.0700184104
Utilizing the activation mechanism of serine proteases to engineer hepatocyte growth factor into a Met antagonist
Abstract
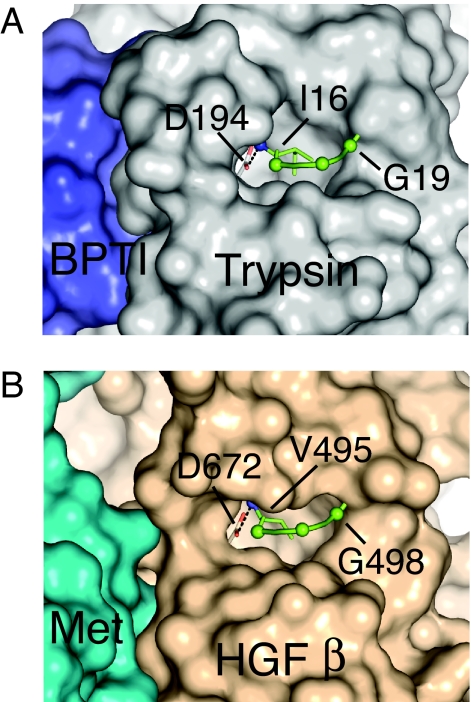
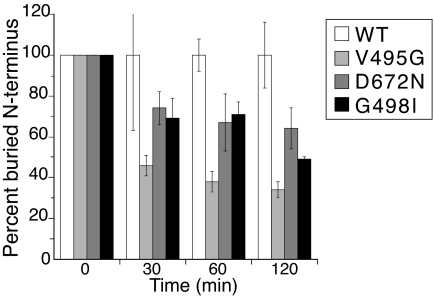
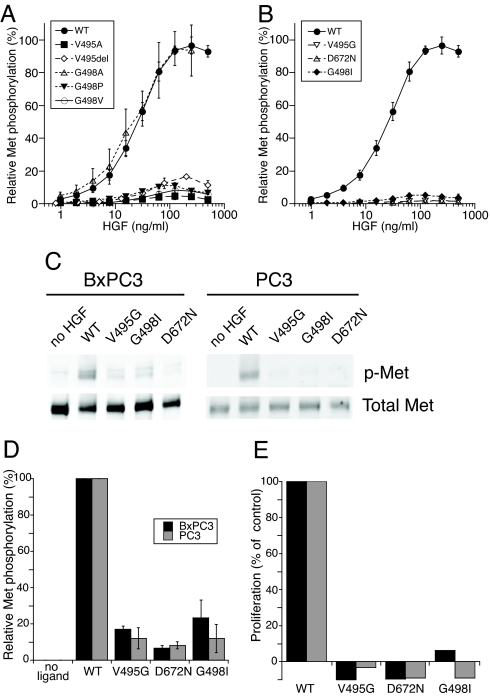
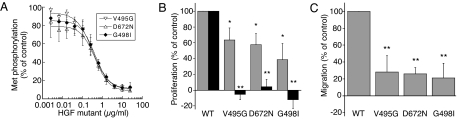
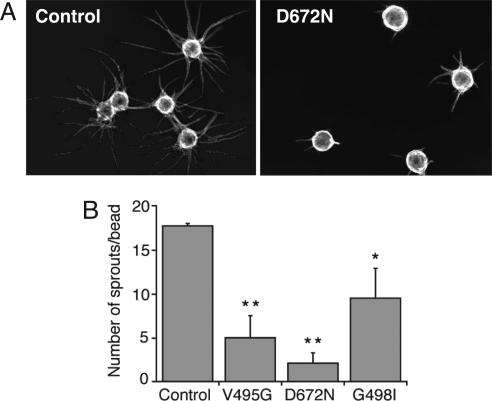
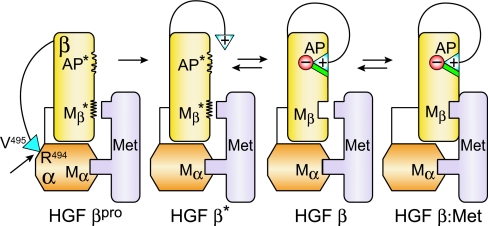
Hepatocyte growth factor (HGF), the ligand for the receptor tyrosine kinase Met, is secreted as single chain pro-HGF that lacks signaling activity. Pro-HGF acquires functional competence upon cleavage between R494 and V495, generating a disulfide-linked alpha/beta-heterodimer, where the beta-chain of HGF (HGF beta) has a serine protease fold that lacks enzymatic activity. We show that, like serine proteases, insertion of the newly formed N terminus in the beta-chain is critical for activity, here by allosterically stabilizing interactions with Met. The HGF beta crystal structure shows that V495 inserts into the "activation pocket" near the Met binding site where the positively charged N terminus forms a salt bridge with the negatively charged D672, and the V495 side chain has hydrophobic interactions with main- and side-chain residues. Full-length two-chain HGF mutants designed to interrupt these interactions (D672N, V495G, V495A, G498I, and G498V) displayed <10% activity in Met receptor phosphorylation, cell migration, and proliferation assays. Impaired signaling of full-length mutants correlated with >50-fold decreases in Met binding of the low-affinity HGF beta domain alone bearing the same mutations and further correlated with impaired N-terminal insertion. Because high-affinity binding resides in the HGF alpha-chain, full-length mutants maintained normal Met binding and efficiently inhibited HGF-mediated Met activation. Conversion of HGF from agonist to antagonist was achieved by as little as removal of two methyl groups (V495A) or a single charge (D672N). Thus, although serine proteases and HGF have quite distinct functions in proteolysis and Met signal transduction, respectively, they share a similar activation mechanism.
Conflict of interest statement
Conflict of interest statement: The authors of this work are employed by Genentech, Inc.
Figures
Similar articles
-
Noncompetitive inhibition of hepatocyte growth factor-dependent Met signaling by a phage-derived peptide.J Mol Biol. 2009 Jan 9;385(1):79-90. doi: 10.1016/j.jmb.2008.09.091. Epub 2008 Oct 15. J Mol Biol. 2009. PMID: 18973760
-
Structure, biosynthesis and biochemical properties of the HGF receptor in normal and malignant cells.EXS. 1993;65:131-65. EXS. 1993. PMID: 8380735 Review.
-
Structural and functional basis of the serine protease-like hepatocyte growth factor beta-chain in Met binding and signaling.J Biol Chem. 2004 Sep 17;279(38):39915-24. doi: 10.1074/jbc.M404795200. Epub 2004 Jun 24. J Biol Chem. 2004. PMID: 15218027
-
Hepatocyte growth factor and its receptor, the tyrosine kinase encoded by the c-MET proto-oncogene.Cell Mol Biol (Noisy-le-grand). 1994 Jul;40(5):597-604. Cell Mol Biol (Noisy-le-grand). 1994. PMID: 7981617 Review.
-
Allosteric peptide activators of pro-hepatocyte growth factor stimulate Met signaling.J Biol Chem. 2010 Dec 17;285(51):40362-72. doi: 10.1074/jbc.M110.179721. Epub 2010 Oct 11. J Biol Chem. 2010. PMID: 20937841 Free PMC article.
Cited by
-
HGF-MET cascade, a key target for inhibiting cancer metastasis: the impact of NK4 discovery on cancer biology and therapeutics.Int J Mol Sci. 2013 Jan 7;14(1):888-919. doi: 10.3390/ijms14010888. Int J Mol Sci. 2013. PMID: 23296269 Free PMC article.
-
Hepsin enhances liver metabolism and inhibits adipocyte browning in mice.Proc Natl Acad Sci U S A. 2020 Jun 2;117(22):12359-12367. doi: 10.1073/pnas.1918445117. Epub 2020 May 13. Proc Natl Acad Sci U S A. 2020. PMID: 32404422 Free PMC article.
-
Direct binding of hepatocyte growth factor and vascular endothelial growth factor to CD44v6.Biosci Rep. 2015 Jun 29;35(4):e00236. doi: 10.1042/BSR20150093. Biosci Rep. 2015. PMID: 26181364 Free PMC article.
-
c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase.Oncogene. 2011 Apr 28;30(17):2003-16. doi: 10.1038/onc.2010.586. Epub 2011 Jan 10. Oncogene. 2011. PMID: 21217780 Free PMC article.
-
Targeting the HGF/Met signalling pathway in cancer.Eur J Cancer. 2010 May;46(7):1260-70. doi: 10.1016/j.ejca.2010.02.028. Epub 2010 Mar 19. Eur J Cancer. 2010. PMID: 20303741 Free PMC article. Review.
References
-
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Nat Rev Mol Cell Biol. 2003;4:915–925. - PubMed
-
- Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Crit Rev Oncol Hematol. 2005;53:35–69. - PubMed
-
- Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Cytokine Growth Factor Rev. 2002;13:41–59. - PubMed
-
- Trusolino L, Comoglio PM. Nat Rev Cancer. 2002;2:289–300. - PubMed
-
- Christensen JG, Burrows J, Salgia R. Cancer Lett. 2005;225:1–26. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
