

Different telomere damage signaling pathways in human and mouse cells
- PMID: 12169636
- PMCID: PMC126171
- DOI: 10.1093/emboj/cdf433
Different telomere damage signaling pathways in human and mouse cells
Abstract
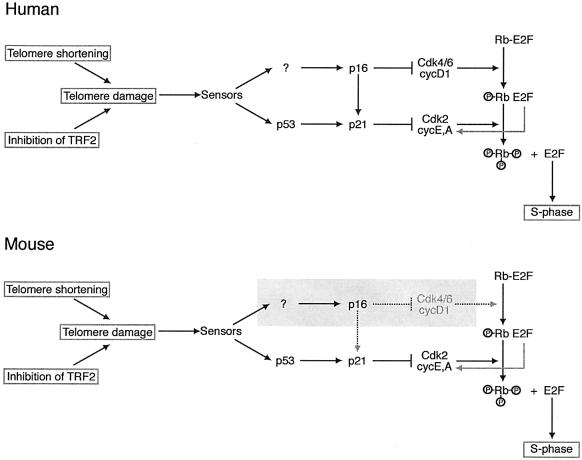
Programmed telomere shortening in human somatic cells is thought to act as a tumor suppressor pathway, limiting the replicative potential of developing tumor cells. Critically short human telomeres induce senescence either by activating p53 or by inducing the p16/RB pathway, and suppression of both pathways is required to suppress senescence of aged human cells. Here we report that removal of TRF2 from human telomeres and the ensuing de-protection of chromosome ends induced immediate premature senescence. Although the telomeric tracts remained intact, the TRF2(DeltaBDeltaM)-induced premature senescence was indistinguishable from replicative senescence and could be mediated by either the p53 or the p16/RB pathway. Telomere de-protection also induced a growth arrest and senescent morphology in mouse cells. However, in this setting the loss of p53 function was sufficient to completely abrogate the arrest, indicating that the p16/RB response to telomere dysfunction is not active in mouse cells. These findings reveal a fundamental difference in telomere damage signaling in human and mouse cells that bears on the use of mouse models for the telomere tumor suppressor pathway.
Figures
Similar articles
-
Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a).Mol Cell. 2004 May 21;14(4):501-13. doi: 10.1016/s1097-2765(04)00256-4. Mol Cell. 2004. PMID: 15149599
-
Significant role for p16INK4a in p53-independent telomere-directed senescence.Curr Biol. 2004 Dec 29;14(24):2302-8. doi: 10.1016/j.cub.2004.12.025. Curr Biol. 2004. PMID: 15620660
-
Loss of p16(Ink4a) function rescues cellular senescence induced by telomere dysfunction.Int J Mol Sci. 2012;13(5):5866-5877. doi: 10.3390/ijms13055866. Epub 2012 May 16. Int J Mol Sci. 2012. PMID: 22754337 Free PMC article.
-
Senescence and immortalization of human cells.Biogerontology. 2000;1(2):103-21. doi: 10.1023/a:1010000132671. Biogerontology. 2000. PMID: 11707927 Review.
-
Genes involved in the control of cellular proliferative potential.Ann N Y Acad Sci. 1998 Nov 20;854:8-19. doi: 10.1111/j.1749-6632.1998.tb09887.x. Ann N Y Acad Sci. 1998. PMID: 9928415 Review.
Cited by
-
Association of telomere instability with senescence of porcine cells.BMC Cell Biol. 2012 Dec 15;13:36. doi: 10.1186/1471-2121-13-36. BMC Cell Biol. 2012. PMID: 23241441 Free PMC article.
-
TGF-beta receptor mediated telomerase inhibition, telomere shortening and breast cancer cell senescence.Protein Cell. 2017 Jan;8(1):39-54. doi: 10.1007/s13238-016-0322-1. Epub 2016 Sep 30. Protein Cell. 2017. PMID: 27696331 Free PMC article.
-
Telomere C-Strand Fill-In Machinery: New Insights into the Human CST-DNA Polymerase Alpha-Primase Structures and Functions.Subcell Biochem. 2024;104:73-100. doi: 10.1007/978-3-031-58843-3_5. Subcell Biochem. 2024. PMID: 38963484 Review.
-
Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells.Cancer Cell. 2012 Jun 12;21(6):765-76. doi: 10.1016/j.ccr.2012.03.044. Cancer Cell. 2012. PMID: 22698402 Free PMC article.
-
Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities.Protein Sci. 2005 Jan;14(1):119-30. doi: 10.1110/ps.04983705. Protein Sci. 2005. PMID: 15608118 Free PMC article.
References
-
- Artandi S.E., Chang,S., Lee,S.L., Alson,S., Gottlieb,G.J., Chin,L. and DePinho,R.A. (2000) Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature, 406, 641–645. - PubMed
-
- Blasco M.A., Lee,H.W., Hande,M.P., Samper,E., Lansdorp,P.M., DePinho,R.A. and Greider,C.W. (1997) Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell, 91, 25–34. - PubMed
-
- Bodnar A.G. et al. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. - PubMed
-
- Broccoli D., Smogorzewska,A., Chong,L. and de Lange,T. (1997) Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet., 17, 231–235. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
